Characterization of developmental and molecular factors underlying release heterogeneity at Drosophila synapses
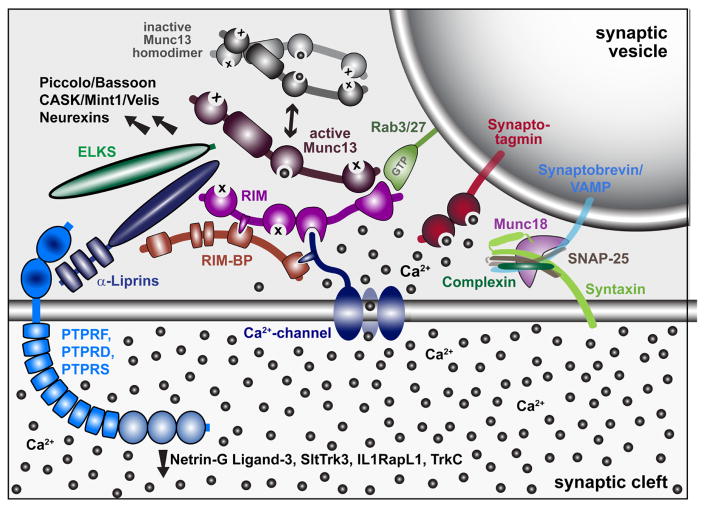
Neurons communicate through neurotransmitter release at specialized synaptic regions known as active zones (AZs). Using biosensors to visualize single synaptic vesicle fusion events at Drosophila neuromuscular junctions, this study analyzed the developmental and molecular determinants of release probability (Pr) for a defined connection with ~300 AZs. Pr was heterogeneous but represented a stable feature of each AZ. Pr remained stable during high frequency stimulation and retained heterogeneity in mutants lacking the Ca(2+) sensor Synaptotagmin 1. Pr correlated with both presynaptic Ca(2+) channel abundance and Ca(2+) influx at individual release sites. Pr heterogeneity also correlated with glutamate receptor abundance, with high Pr connections developing receptor subtype segregation. Intravital imaging throughout development revealed that AZs acquire high Pr during a multi-day maturation period, with Pr heterogeneity largely reflecting AZ age. The rate of synapse maturation was activity-dependent, as both increases and decreases in neuronal activity modulated glutamate receptor field size and segregation (Akbergenova, 2018).
This study used quantal imaging, super resolution SIM, and intravital imaging to examine the development of heterogeneity in evoked Pr across the AZ population at Drosophila NMJs. It was first confirmed that release heterogeneity was not caused by summation of fusion events from multiple unresolvable AZs. Indeed, high Pr sites corresponded to single AZs with enhanced levels of BRP. These findings are consistent with previous observations using conventional light microscopy that indicate Pr correlates with BRP levels. By monitoring release over intervals of extensive vesicle fusion during strong stimulation, it was also observed that Pr is a stable feature of each AZ. In addition, loss of the synaptic vesicle Ca2+ sensor Syt1 globally reduced Pr without altering the heterogeneous distribution of Pr across AZs, indicating that AZ-local synaptic vesicle pools with differential Ca2+ sensitivity are not likely to account for Pr heterogeneity. Since voltage gated calcium channel (VGCC) abundance, gating, and organization within the AZ are well established regulators of Pr across synapses, heterogeneity in presynaptic Ca2+ channel abundance was a clear candidate for the generation of Pr heterogeneity at Drosophila NMJs. Indeed, the Cac Ca2+ channel responsible for neurotransmitter release is heterogeneously distributed across the NMJ . Using transgenically labeled Cac lines, it was observed that Cac density at AZs is indeed strongly correlated with Pr. To directly visualize presynaptic Ca2+ influx at single AZs, GCaMP fusions were generated to the core AZ component BRP. Ca2+ influx at single AZs was highly correlated with both Cac density and Pr. The cacNT27 mutant with decreased conductance also resulted in a global reduction in Pr without disrupting heterogeneity, further confirming that Ca2+ influx regulates Pr across the range of release heterogeneity (Akbergenova, 2018).
Postsynaptically, high Pr AZs were enriched in GluRIIA-containing receptors and displayed a distinct pattern of glutamate receptor clustering. While most synapses showed GluRIIA and GluRIIB spread over the entire PSD, high Pr AZs were apposed by PSDs where GluRIIA concentrated at the center of the AZ, with GluRIIB forming a ring at the PSD periphery. Indeed, anti-glutamate receptor antibody staining of wildtype larvae lacking tagged glutamate receptors had previously identified a GluRIIB ring around the GluRIIA core in some mature third instar NMJ AZs (Marrus , 2004). In addition, activity-dependent segregation of GluRIIA and a GluRIIA gating mutant has been observed at individual AZs in Drosophila (Petzoldt, 2014). The correlation of Pr with GluRIIA accumulation is especially intriguing considering that this subunit has been implicated in homeostatic and activity-dependent plasticity (Davis, 2006; Frank, 2014; Petersen et al., 1997; Sigrist et al., 2003). By following the developmental acquisition of this postsynaptic property as a proxy for Pr from the first through third instar larval stages via intravital imaging in control and mutant backgrounds, it was observed that the earliest formed AZs are the first to acquire this high Pr signature over a time course of ~3 days, and that PSD maturation rate can be modulated by changes in presynaptic activity (Akbergenova, 2018).
Similar to prior observations, this study found that most AZs at the Drosophila NMJ have a low Pr. For the current study, the AZ pool was artificially segregated into low and high release sites, with high releasing sites defined based on having a release rate greater than two standard deviations above the mean. Given that birthdate is a key predictor of glutamate receptor segregation, and by proxy Pr, the AZ pool is expected to actually reflect a continuum of Pr values based on their developmental history. However, using the two standard deviation criteria, 9.9% of AZs fell into the high Pr category, with an average Pr of 0.28. It was also observed that 9.7% of the AZs analyzed displayed only spontaneous release. No fusion events could be detected for either evoked or spontaneous release for another 14.6% of AZs that were defined by a GluRIIA-positive PSD in live imaging. Future investigation will be required to determine whether these cases represent immature AZs with extremely low evoked Pr, or distinct categories reflective of differences in AZ content. The remaining AZs that participated in evoked release had an average Pr of 0.05. Ca2+ channel density and Ca2+ influx at individual AZs was a key determinant of evoked Pr heterogeneity, as Pr and the intensity of Cac channels tagged with either TdTomato or GFP displayed a strong positive correlation. Spontaneous fusion showed a much weaker correlation with both Cac density and Ca2+ influx at individual AZs, consistent with prior studies indicating spontaneous release rates are poorly correlated with external Ca2+ levels at this synapse. With synaptic vesicle fusion showing a steep non-linear dependence upon external Ca2+ with a slope of ~3-4, a robust change in Pr could occur secondary to a relatively modest increase in Ca2+ channel abundance over development. Although the number of VGCCs at a Drosophila NMJ AZ is unknown, estimates of Cac-GFP fluorescence during quantal imaging indicate a ~ 2 fold increase in channel number would be necessary to move a low Pr AZ into the high Pr category. Similar correlations between evoked Pr and Ca2+ channel abundance have been found at mammalian synapses, suggesting this represents a common evolutionarily conserved mechanism for determining release strength at synapses (Akbergenova, 2018).
This study did not test the correlation of Pr with other AZ proteins besides Cac and BRP, but it would not be surprising to see a positive correlation with the abundance of many AZ proteins based on the observation that maturation time is a key determinant for Pr. Indeed, recent studies have begun to correlate Pr with specific AZ proteins at Drosophila NMJs. It was also observed that PSD size was robustly increased by 1.6-fold over a 24 hr period of AZ development during the early larval period in control animals. AZ maturation is likely to promote increased synaptic vesicle docking and availability, consistent with observations that correlate AZ size with either Pr or the readily releasable pool (Akbergenova, 2018).
Several models are considered for how AZs acquire this heterogeneous nature of Pr distribution during a developmental period lasting several days. One possibility is that unique AZs gain high Pr status through a mechanism that would result in preferential accumulation of key AZ components compared to their neighbors. Given that retrograde signaling from the muscle is known to drive synaptic development at Drosophila NMJs, certain AZ populations might have preferential access to specific signaling factors that would alter their Pr state. Another model is that AZs compete for key presynaptic Pr regulators through an activity-dependent process. High Pr AZs might also be more mature than their low Pr neighbors, having a longer timeframe to accumulate AZ components. Given the Drosophila NMJ is constantly forming new AZs at a rapid pace during development, newly formed AZs would be less mature compared to a smaller population of 'older' high Pr AZs (Akbergenova, 2018).
To examine if the release heterogeneity observed at the third instar stage reflects AZ birth order over several days of development, the intravital imaging was extended through a longer time period beginning in the first instar larval stage. GCaMP imaging indicated high Pr sites segregate GluRIIA and GluRIIB differently from low Pr sites, with the IIA isoform preferentially localizing at the center of PSDs apposing high Pr AZs. As such, developmental acquisition of this property was used as an indicator of high Pr sites. Although segregation of glutamate receptors may not perfectly replicate the timing of Pr acquisition during development, it is currently the best tool for estimating Pr during sequential live imaging. Based on the acquisition of GluRIIA/B segregation, the data support the hypothesis that increases in Pr reflect a time-dependent maturation process at the NMJ. The continuous addition of new AZs, which double in number during each day of development, ensures that the overall ratio of high to low Pr sites represents a low percentage as the NMJ grows. Confidence in the age-dependency model of Pr was further established by mapping release after following NMJs intravitally for 24 hr; using this approach, it was found that newly formed AZs are consistently very low Pr. Finally, Pr was mapped in the second instar stage, and it was observed that the heterogeneity at this stage is shifted towards higher releasing sites, with a reduction in the fraction of low-releasing sites (Akbergenova, 2018).
These data indicate AZs are born with low Pr and gain pre- and postsynaptic material over 3 days on an upward trajectory toward higher Pr status. Is AZ age a static determinant of Pr, or can growth rate be regulated to allow faster or slower acquisition of high Pr AZs? To answer this question, whether mutants that alter presynaptic activity influence the rate of PSD growth and GluRIIA/B segregation was assayed. In BRP69/def, syt1null, and napTS mutants with decreased presynaptic release, a significant reduction in postsynaptic maturation rate and in the percentage of PSDs with GluRIIA/IIB rings was observed. Conversely, in the shaker, eag double mutant with increased presynaptic excitability, a significantly increased rate of GluRIIB field size and a significant increase in GluRIIA/IIB rings was found compared to controls. To investigate whether differences in synapse growth and maturation rate could be seen between AZs enriched in BRP and Cac versus neighboring AZs that are deficient in these components, development was imaged in the rab3 null mutant. At the first instar stage when AZs are roughly age matched, release sites enriched in presynaptic components had developed large and mature PSDs in stark contrast with non-enriched AZs, whose PSDs appeared immature. These results indicate that PSD maturation can be influenced by presynaptic activity at the resolution of single AZs (Akbergenova, 2018).
Although how AZs are assembled during development is still being established, the current data do not support a model where AZs are fully preassembled during transport and then deposited as a single 'quantal' entity onto the presynaptic membrane. Rather, these data support a model of seeding of AZ material that increases developmentally over time as AZs matures, consistent with previous studies of AZ development in Drosophila. Although no evidence for rapid changes in Pr were detected in the steady-state conditions used in the current study, homeostatic plasticity is known to alter Pr over a rapid time frame (~10 min) at the NMJ. It will be interesting to determine if Cac abundance can change over such a rapid window, or whether the enhanced release is mediated solely through changes in Cac function and Ca2+ influx. Changes in the temporal order of Pr development could also occur secondary to altered transport or capture of AZ material. For example, the large NMJ on muscle fibers 6 and 7 displays a gradient in synaptic transmission, with terminal branch boutons often showing a larger population of higher Pr AZs. If AZ material is not captured by earlier synapses along the arbor, it would be predicted to accumulate in terminal boutons, potentially allowing these AZs greater access to key components, and subsequently increasing their rate of Pr acquisition. Alternatively, the gradient of Pr along the axon could be due to terminal boutons being slightly older than the rest of the arbor (Akbergenova, 2018).
In summary, the current data indicate that heterogeneity in release correlates highly with Ca2+ channel abundance and Ca2+ influx at AZs. Postsynaptically, PSDs apposed to high-releasing AZs display increased GluRIIA abundance and form segregated receptor fields, with GluRIIB forming a ring around a central core of GluRIIA. Release sites accumulate these high Pr markers during a synapse maturation process in which newly formed AZs are consistently low Pr, with AZs gaining signatures of high releasing sites over several days. Finally, mutations that increase or decrease presynaptic activity result in faster or slower rates of PSD maturation, respectively. These data add to the understanding of the molecular and developmental features associated with high versus low Pr AZs (Akbergenova, 2018).
A neuropeptide signaling pathway regulates synaptic growth in Drosophila
Neuropeptide signaling is integral to many aspects of neural communication, particularly modulation of membrane excitability and synaptic transmission. However, neuropeptides have not been clearly implicated in synaptic growth and development. This study demonstrates that cholecystokinin-like receptor (CCK-like receptor at 17D1; CCKLR), and Drosulfakinin (DSK), its predicted ligand, are strong positive growth regulators of the Drosophila melanogaster larval neuromuscular junction (NMJ). Mutations of CCKLR (CCKLR-17D1 but not CCKLR-17D3) or dsk produce severe NMJ undergrowth, whereas overexpression of CCKLR causes overgrowth. Presynaptic expression of CCKLR is necessary and sufficient for regulating NMJ growth. CCKLR and dsk mutants also reduce synaptic function in parallel with decreased NMJ size. Analysis of double mutants revealed that DSK/CCKLR regulation of NMJ growth occurs through the cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA)-cAMP response element binding protein (CREB) pathway. These results demonstrate a novel role for neuropeptide signaling in synaptic development. Moreover, because the cAMP-PKA-CREB pathway is required for structural synaptic plasticity in learning and memory, DSK/CCKLR signaling may also contribute to these mechanisms (Chen, 2012).
Proper synaptic growth is essential for normal development of the nervous system and its function in mediating complex behaviors such as learning and memory. The Drosophila larval neuromuscular junction (NMJ) has become a powerful system for studying the molecular mechanisms underlying synaptic development and plasticity, and many of the key synaptic proteins are evolutionarily conserved (Chen, 2012).
Genetic and molecular analysis in Drosophila has uncovered numerous molecules and pathways that regulate NMJ growth, including proteins required for cell adhesion, endocytosis, cytoskeletal organization, and signal transduction via TGF-β, Wingless, JNK, cAMP, and other signaling molecules. For example, previous studies revealed that increased cAMP levels led to a down-regulation of the cell adhesion protein FasII at synapses, and the activation of the cAMP response element binding protein (CREB) transcription factor to achieve long-lasting changes in synaptic structure and function. Despite the identification and characterization of these various positive and negative regulators, understanding of the networks that govern synaptic growth is still incomplete, with many of the key components and mechanisms yet to be uncovered and analyzed (Chen, 2012).
To search for new regulators of synaptic growth, a forward genetic screen was conducted for mutants exhibiting altered NMJ morphology. In this screen, a new mutation was discovered that exhibits strikingly undergrown NMJs, which indicates disruption of a positive regulator of NMJ growth. The affected protein was identified as cholecystokinin (CCK)-like receptor (CCKLR), a putative neuropeptide receptor that belongs to the family of G-protein coupled receptors (GPCRs) sharing a uniform topology with seven transmembrane domains. When activated by their ligands, neuropeptide GPCRs affect levels of second messengers such as cAMP, diacylglycerol, inositol trisphosphate, and intracellular calcium. Through activation of their cognate receptors, secreted neuropeptides mediate communication among various sets of neurons as well as other cell types to regulate several physiological activities, including feeding and growth, molting, cuticle tanning, circadian rhythm, sleep, and learning and memory. In general, neuropeptides act by modulating neuronal activity through both short-term and long-term effects. Short-term effects include modifications of ion channel activity and alterations in release of or response to neurotransmitters. Long-term effects include changes in gene expression through activation of transcription factors and protein synthesis. In contrast with the well-known effects of neuropeptide signaling on neuronal activity and the strength of synaptic transmission, regulation of synaptic growth and development by neuropeptides has not previously been clearly established (Chen, 2012).
This study demonstrates that CCKLR is required presynaptically to promote NMJ growth. Moreover, mutations of drosulfakinin
(dsk), which encode the predicted ligand of CCKLR, cause similar NMJ undergrowth and interact genetically with CCKLR mutations, indicating that DSK and CCKLR are components of a common signaling mechanism that regulates NMJ growth. In addition to the morphological phenotypes caused by mutations of CCKLR and dsk, mutant larvae also exhibit deficits in synaptic function. Through phenotypic analysis of double mutant combinations, it was shown that DSK/CCKLR signals through the cAMP-PKA-CREB pathway to regulate NMJ growth. The results suggest a novel role for neuropeptide signaling in regulation of synaptic development. Moreover, because the cAMP-PKA-CREB pathway is required for structural plasticity of synapses in learning and memory, DSK/CCKLR signaling may also contribute to these mechanisms (Chen, 2012).
Neuropeptides, whose effects have been extensively studied at NMJs, are usually described as neuromodulators because they modify the strength of synaptic transmission. For example, proctolin can potentiate the action of glutamate at certain NMJs in insects. However, involvement of neuropeptides in regulating neural development has not been well characterized. Recently, a C-type natriuretic peptide acting through a cGMP signaling cascade was found to be required for sensory axon bifurcation in mice, which suggests that neuropeptides may have a broader role in development than previously appreciated. The current studies demonstrate that DSK and its receptor, CCKLR, are strong positive regulators of NMJ growth in Drosophila (Chen, 2012).
DSK belongs to the family of FMRFamide-related peptides (FaRPs), which is very broadly distributed across invertebrate and vertebrate phyla. Originally identified in clams, FaRPs affect heart rate, blood pressure, gut motility, feeding behavior, and reproduction in invertebrates. They have been shown to enhance synaptic efficacy at NMJs in locust and to modulate presynaptic Ca2+ channel activity in crustaceans. In Drosophila, various neuropeptides derived from the FMRFa gene can modulate the strength of muscle contraction when perfused onto standard larval nerve-muscle preparations. To these previously described functions of FaRPs, this study adds a new role as a positive regulator of NMJ development (Chen, 2012).
Transgenic rescue experiments, RNAi expression, and overexpression of WT CCKLR demonstrate that CCKLR functions presynaptically in motor neurons to promote NMJ growth. Downstream components of this pathway were identified on the basis of known biochemistry of GPCRs and phenotypic interactions in double mutant combinations. GPCRs typically function by activating second messenger pathways via G proteins. Because loss-of-function mutations in dgs (which encodes the Gsα subunit in Drosophila) cause NMJ undergrowth, it is hypothesized that CCKLR signals through Gsα. Consistent with this idea, it was found that presynaptic constitutively active dgs overexpression rescues the NMJ undergrowth phenotype of CCKLR mutants. Conversely, dominant dose-dependent interactions were observed between CCKLR-null mutations and mutations of rut, which encodes an AC; or PKA-C1, which encodes a cAMP-dependent protein kinase, resulting in significant reductions in NMJ growth. These data place CCKLR together with the other genes in a common cAMP-dependent signaling pathway that regulates NMJ growth (Chen, 2012).
It is known that the AC encoded by rut is activated by Gsα, and on the basis of the results, it is proposed that Gsα is downstream of CCKLR signaling. However, the NMJ undergrowth in rut1, which is a presumptive null mutant, is not as severe as that of a CCKLR-null mutant. This is likely due to the fact that the Drosophila genome contains up to seven different AC-encoding genes, all of which are stimulated by Gsα. Presumably, one or more additional AC-encoding genes share some functional overlap with rut in regulation of NMJ growth. This idea is in good agreement with the results of Wolfgang (2004), who found that the NMJ undergrowth phenotype of rut1 is weaker than that of dgs mutants, which they also interpreted as an indication that multiple ACs are activated by the Gsα encoded by dgs (Chen, 2012).
The primary effector of this pathway is CREB2, a transcriptional regulatory protein that is activated upon phosphorylation by PKA. Consistent with the idea that CCKLR ultimately acts via activation of CREB, loss-of-function mutations of dCreb2 or neuronal overexpression of a dominant-negative dCreb2 transgene cause NMJ undergrowth similar to that of CCKLR-null mutants. Additionally, loss of one copy of dCreb2 in a CCKLR heterozygous background also causes NMJ undergrowth, and overexpression of WT dCreb2 fully rescues the NMJ undergrowth phenotype of CCKLR null, even leading to NMJ overgrowth. Thus, regulation of NMJ growth through the CCKLR signaling pathway is clearly mediated by dCreb2, whose activity is itself necessary and sufficient for regulating NMJ growth. This conclusion differs from another study that suggested that dCreb2 is required for NMJ function but not NMJ growth. One possible explanation for this discrepancy is that a weaker, inducible heat shock-driven transgene was used to express dCreb2 in the earlier work, whereas strong constitutive neuronal drivers were used this study. In any case, the current results demonstrate that in addition to its known role in NMJ function, CREB2 is also a strong positive regulator of NMJ growth and is likely to play a greater role in structural plasticity of synapses in learning and memory in Drosophila than previously suggested. This conclusion is consistent with a recent study indicating that sprouting of type II larval NMJs in response to starvation is stimulated by a cAMP/CREB-dependent pathway via activation of an octopamine GPCR (Chen, 2012).
In addition to being undergrown, NMJs in CCKLR mutant larvae also exhibit a functional deficit. This is perhaps less straightforward than it might seem. Previous analyses of mutations affecting growth of the larval NMJ in Drosophila have shown that there is no simple correlation between the size and complexity of the NMJ and the amplitude of EJPs or amount of neurotransmitter release. These discrepancies arise because of various homeostatic compensatory mechanisms and because some of the affected signaling pathways alter synaptic growth and synaptic function in different ways via distinct downstream targets. For example, a mutation in highwire, which has the most extreme NMJ overgrowth phenotype described, is associated with a decrease in synaptic transmission. Wallenda mutations have been shown to fully suppress the overgrowth phenotype, but have no effect on the deficit in synaptic transmission (Chen, 2012).
In the case of CCKLR mutants, however, there appears to be a very good correspondence between the morphological phenotype and the electrophysiological phenotype: the reduction in the total number of active zones in CCKLR larvae as measured morphologically correlates very well with the reduction in quantal content that was observe. In addition, no difference in CCKLR mutants was detected in calcium sensitivity of transmitter release or in the size or frequency of spontaneous release events. Thus, the synaptic growth phenotype of CCKLR mutant NMJs is sufficient to account for the functional phenotype. However, the possibility cannot be ruled out that DSK/CCKLR signaling also exerts some modulatory effect on NMJ function that is distinct from its effect on NMJ development (Chen, 2012).
DSK is identified as the Drosophila orthologue of CCK, the ligand of CCKLR in vertebrates, on the basis of sequence analysis. The genetic analysis strongly supports the conclusion that DSK is the ligand of CCKLR at the larval NMJ. First, mutations of dsk and expression of dsk RNAi result in NMJ undergrowth phenotypes similar to that of CCKLR mutants. Second, loss of one copy of both dsk and CCKLR in double heterozygotes results in NMJ undergrowth. Third, heterozygosity for dsk does not further enhance the phenotype of a CCKLR-null mutant as expected if DSK regulates NMJ growth through its action on CCKLR. Fourth, overexpression of UAS-dsk does not rescue the undergrowth phenotype of a CCKLR-null mutant, but CCKLR overexpression can rescue NMJ undergrowth of a dsk hypomorphic mutant (Chen, 2012).
The discovery of an entirely novel role for neuropeptide signaling in NMJ growth raises several questions about how this signaling is regulated and the biological significance of this mechanism. Although answers to these questions will require much additional work, an immediate question is whether a paracrine or autocrine mechanism is involved. In the case of octopamine-mediated synaptic sprouting in response to starvation, both autocrine and paracrine signaling are involved in the sprouting of type II and type I NMJs, respectively. In an early immunohistochemical investigation, it was reported that DSK was detected in medial neurosecretory cells in the larval CNS that extended projections anteriorly into the brain and posteriorly to the ventral ganglion. As it was not possible to obtain the original DSK antiserum and raising a new antiserum was not successful, the previous report has not been extended or confirmed. Instead, tissue-specific RNAi experiments were performed to examine the spatial requirement for DSK. Pan-neuronal dsk RNAi expression indicates that DSK expression in neurons is required to promote NMJ growth. In addition, C739-Gal4-driven dsk RNAi also causes NMJ undergrowth, whereas OK-Gal4-driven dsk RNAi in motor neurons does not. The expression pattern of C739-Gal4 overlaps with the DSK-positive cells previously identified by immunohistochemistry, which suggests that DSK produced by those neurosecretory cells is required for normal NMJ growth. Thus, from available data, it seems most likely that DSK is acting in paracrine fashion to regulate NMJ growth. However, further investigation will be necessary to determine the exact source of the DSK that promotes NMJ growth to fully understand how this neuropeptide regulates NMJ development (Chen, 2012).
Studies have demonstrated a role for CREB in long-term synaptic plasticity-structural changes in synaptic morphology that underlie the formation of long-term memories. This study shows that in addition to CREB's role in structural modification of synapses in response to experience after development is complete, it is also a key regulator of growth and morphology during development of the larval NMJ. Moreover, although CREB is the transcriptional effector for many GPCRs, the fact that NMJs in CCKLR mutants are as undergrown as those of CREB mutants suggests that DSK/CCKLR signaling is a major input to CREB during NMJ growth. Many of the genes encoding intermediate components of the pathway such as dnc, rut, and PKA also have effects on NMJ growth and development as well as on synaptic plasticity and learning and memory, further emphasizing an overlap between the mechanisms that regulate synaptic growth during development and those that regulate postdevelopmental structural synaptic plasticity. These results raise the possibility that DSK/CCKLR signaling also plays a role in long-term synaptic plasticity and learning as well as in synaptic development (Chen, 2012).
Heterotrimeric Go protein links Wnt-Frizzled signaling with ankyrins to regulate the neuronal microtubule cytoskeleton
Drosophila neuromuscular junctions (NMJs) represent a powerful model system with which to study glutamatergic synapse formation and remodeling. Several proteins have been implicated in these processes, including components of canonical Wingless (Drosophila Wnt1) signaling and the giant isoforms of the membrane-cytoskeleton linker Ankyrin 2, but possible interconnections and cooperation between these proteins were unknown. This study demonstrates that the heterotrimeric G protein Go functions as a transducer of Wingless-Frizzled 2 signaling in the synapse. Ankyrin 2 was identified as a target of Go signaling required for NMJ formation. Moreover, the Go-ankyrin interaction is conserved in the mammalian neurite outgrowth pathway. Without ankyrins, a major switch in the Go-induced neuronal cytoskeleton program is observed, from microtubule-dependent neurite outgrowth to actin-dependent lamellopodial induction. These findings describe a novel mechanism regulating the microtubule cytoskeleton in the nervous system. This work in Drosophila and mammalian cells suggests that this mechanism might be generally applicable in nervous system development and function (Luchtenborg, 2014).
Ankyrins (Ank) are highly abundant modular proteins that mediate protein-protein interactions, mainly serving as adaptors for linking the cytoskeleton to the plasma membrane. Mammalian genomes encode three Ank genes [AnkR (Ank1), AnkB (Ank2) and AnkG (Ank3)], whereas Drosophila has two [Ank1 (also known as Ank - FlyBase) and Ank2]. Ank2 is expressed exclusively in neurons and exists in several splicing variants. The larger isoforms (Ank2M, Ank2L and Ank2XL) are localized to axons and play important roles in NMJ formation and function. The C-terminal part of Ank2L can bind to microtubules. Despite the well-established role of Ank2 in NMJ formation, its function has been considered somewhat passive and its mode of regulation has not been clarified. This study shows that Gαo binds to Ank2 and that these proteins and the Wg pathway components Wg, Fz2, and Sgg jointly coordinate the formation of the NMJ. The functional Gαo-Ank interaction is conserved from insects to mammals (Luchtenborg, 2014).
Synaptic plasticity underlies learning and memory. Both in invertebrates and vertebrates, activation of Wnt signaling is involved in several aspects of synapse formation and remodeling, and defects in this pathway may be causative of synaptic loss and neurodegeneration. Thus, understanding the molecular mechanisms of synaptic Wnt signaling is of fundamental as well as medical importance. The Drosophila NMJ is a powerful model system with which to study glutamatergic synapses, and the Wnt pathway has been widely identified as one of the key regulators of NMJ formation.
This study provides important mechanistic insights into Wnt signal transduction in the NMJ, identifying the heterotrimeric Go protein as a crucial downstream transducer of the Wg-Fz2 pathway in the presynapse. It was further demonstrated that Ank2, a known player in the NMJ, is a target of Gαo in this signaling (Luchtenborg, 2014).
This study found that the α subunit of Go is strongly expressed in the presynaptic cell, and that under- or overactivation of this G protein leads to neurotransmission and behavioral defects. At the level of NMJ morphology, presynaptic downregulation or Ptx-mediated inactivation of Gαo was found to recapitulate the phenotypes obtained by similar silencing of wg and fz2. These data confirm that presynaptic Wg signaling, in addition to the Wg pathway active in the muscle, is crucial for proper NMJ formation, and that Go is required for this process. Furthermore, neuronal Gαo overexpression can rescue the wg and fz2 loss-of-function phenotypes, demonstrating that, as in other contexts of Wnt/Fz signaling, Go acts as a transducer of Wg/Fz2 in NMJ formation. In contrast to its evident function and clear localization in the presynapse, Gαo localization on the muscle side of the synapse is much less pronounced or absent. Unlike Gαo, the main Drosophila Gβ subunit is strongly expressed in both the pre- and postsynapse. Thus, a heterotrimeric G protein other than Go might be involved in the postsynaptic Fz2 transduction, as has been implicated in Fz signaling in some other contexts (Luchtenborg, 2014).
A recent study proposed a role for Gαo downstream of the octopamine receptor Octβ1R. This signaling was proposed to regulate the acute behavioral response to starvation both on type II NMJs (octapaminergic) and on the type I NMJs (glutamatergic) analyzed in this study. In contrast to the current observations, downregulation of Gαo in these NMJs was proposed to increase, rather than decrease, type I bouton numbers. It is suspected that the main reason for the discrepancy lies in the Gal4 lines used. The BG439-Gal4 and C380-Gal4 lines of Koon are poorly characterized and, unlike the well-analyzed pan-neuronal elav-Gal4 and motoneuron-specific OK371-Gal4 and D42-Gal4 driver lines used in the current study, might mediate a more acute expression. In this case, this study reflects the positive role of Gαo in the developmental formation of glutamatergic boutons, as opposed to a role in acute fine-tuning in response to environmental factors as studied by Koon (Luchtenborg, 2014).
Postsynaptic expression of fz2 was found to fully rescue fz2 null NMJs. This study found that presynaptic knockdown of Fz2 (and other components of Wg-Fz2-Gαo signaling) recapitulates fz2 null phenotypes, whereas presynaptic overactivation of this pathway increases bouton numbers; furthermore, presynaptic overexpression of fz2 or Gαo rescues the fz2 nulls, just as postsynaptic overexpression of fz2 does. The current data thus support a crucial role for presynaptic Wg-Fz2-Gαo signaling in NMJ formation. Interestingly, both pre- and postsynaptic re-introduction of Arrow, an Fz2 co-receptor that is normally present both pre- and postsynaptically, as is Fz2 itself, can rescue arrow mutant NMJs. Thus, it appears that the pre- and postsynaptic branches of Fz2 signaling are both involved in NMJ development. A certain degree of redundancy between these branches must exist. Indeed, wild-type levels of Fz2 in the muscle are not sufficient to rescue the bouton defects induced by presynaptic expression of RNAi-fz2, yet overexpression of fz2 in the muscle can restore the bouton integrity of fz2 nulls. One might hypothesize that postsynaptic Fz2 overexpression activates a compensatory pathway - such as that mediated by reduction in laminin A signaling - that leads to restoration in bouton numbers in fz2 mutants. The current data showing that the targeted downregulation of Fz2 in the presynapse is sufficient to recapitulate the fz2 null phenotype underpin the crucial function of presynaptic Fz2 signaling in NMJ formation (Luchtenborg, 2014).
This study found that downregulation of Ank2 produces NMJ defects similar to those of wg, fz2 or Gαo silencing. However, Ank2 mutant phenotypes appear more pronounced, indicating that Wg-Fz2-Gαo signaling might control a subset of Ank2-mediated activities in the NMJ. Ank2 was proposed to play a structural role in NMJ formation, binding to microtubules through its C-terminal region. However, since the C-terminal region was insufficient to rescue Ank2L mutant phenotypes, additional domains are likely to mediate Ank2 function through binding to other proteins. This study demonstrates in the yeast two-hybrid system and in pull-down experiments that the ankyrin repeat region of Ank2 physically binds Gαo, suggesting that the function of Ank2 in NMJ formation might be regulated by Wg-Fz2-Gαo signaling. Indeed, epistasis experiments place Ank2 downstream of Gαo in NMJ formation (Luchtenborg, 2014).
Upon dissociation of the heterotrimeric Go protein by activated GPCRs such as Fz2, the liberated Gαo subunit can signal to its downstream targets both in the GTP- and GDP-bound state (the latter after hydrolysis of GTP and before re-association with Gβγ). The free signaling Gαo-GDP form is predicted to be relatively long lived, and a number of Gαo target proteins have been identified that interact equally well with both of the nucleotide forms of this G protein. In the context of NMJ formation, this study found that Gαo-GTP and -GDP are efficient in the activation of downstream signaling, and identifies Ank2 as a binding partner of Gαo that interacts with both nucleotide forms. The importance of signaling by Gα-GDP released from a heterotrimeric complex by the action of GPCRs has also been demonstrated in recent studies of mammalian chemotaxis, planar cell polarity and cancer (Luchtenborg, 2014).
Gαo[G203T], which largely resides in the GDP-binding state owing to its reduced affinity for GTP, might be expected to act as a dominant-negative. However, in canonical Wnt signaling, regulation of asymmetric cell division as well as in planar cell polarity (PCP) signaling in the wing, Gαo[G203T] displays no dominant-negative activity but is simply silent, whereas in eye PCP signaling this form acts positively but is weaker than other Gαo forms. Biochemical characterization of the mammalian Gαi2[G203T] mutant revealed that it can still bind Gβγ and GTP, but upon nucleotide exchange Gαi2[G203T] fails to adopt the activated confirmation and can further lose GTP. The current biochemical characterization confirms that Gαo[G203T] still binds GTP. Interestingly, Gαi2[G203T] inhibited only a fraction of Gαi2-mediated signaling, suggesting that the dominant-negative effects of the mutant are effector specific. Thus, it is inferred that a portion of Gαo[G203T] can form a competent Fz2-transducing complex, and a portion of overexpressed Gαo[G203T] resides in a free GDP-loaded form that is also competent to activate downstream targets - Ank2 in the context of NMJ formation (Luchtenborg, 2014).
These experiments place Ank2 downstream of Gαo and also of Sgg (GSK3β). It remains to be investigated whether Ank2 can directly interact with and/or be phosphorylated by Sgg. Meanwhile, it is proposed that the microtubule-binding protein Futsch might be a linker between Sgg and Ank2. Futsch is involved in NMJ formation and is placed downstream of Wg-Sgg signaling, being the target of phosphorylation and negative regulation by Sgg as the alternative target to β-catenin, which is dispensable in Wg NMJ signaling. Abnormal Futsch localization has been observed in Ank2 mutants. In Drosophila wing and mammalian cells in culture, Gαo acts upstream of Sgg/GSK3β. Cumulatively, these data might suggest that the Wg-Fz2-Gαo cascade sends a signal to Futsch through Sgg, parallel to that mediated by Ank2 (Luchtenborg, 2014).
The importance of the Gαo-Ank2 interaction for Drosophila NMJ development is corroborated by findings in mammalian neuronal cells, where it was demonstrated that the ability of Gαo to induce neurite outgrowth is critically dependent on AnkB and AnkG. Knockdown of either or both ankyrin reduces neurite production. Remarkably, upon AnkB/G downregulation, Gαo switches its activity from the induction of microtubule-dependent processes (neurites) to actin-dependent protrusions (lamellopodia). Furthermore, Gαo recruits AnkB to the growing neurite tips. These data demonstrate that the Gαo-ankyrin mechanistic interactions are conserved from insects to mammals and are important for control over the neuronal tubulin cytoskeleton in the context of neurite growth and synapse formation. The novel signaling mechanism that were uncovered might thus be of general applicability in animal nervous system development and function (Luchtenborg, 2014).
Acetylated alpha-tubulin K394 regulates microtubule stability to shape the growth of axon terminals
Microtubules are essential to neuron shape and function. Acetylation of tubulin has the potential to directly tune the behavior and function of microtubules in cells. Although proteomic studies have identified several acetylation sites in α-tubulin, the effects of acetylation at these sites remains largely unknown. This includes the highly conserved residue lysine 394 (K394), which is located at the α-tubulin dimer interface. Using a fly model, this study showed that α-tubulin K394 is acetylated in the nervous system and is an essential residue. Acetylation-blocking mutation in endogenous α-tubulin, K394R, was found to perturb the synaptic morphogenesis of motoneurons and reduces microtubule stability. Intriguingly, the K394R mutation has opposite effects on the growth of two functionally and morphologically distinct motoneurons, revealing neuron-type-specific responses when microtubule stability is altered. Eliminating the deacetylase HDAC6 increases K394 acetylation, and the over-expression of HDAC6 reduces microtubule stability similar to the K394R mutant. Thus, these findings implicate α-tubulin K394 and its acetylation in the regulation of microtubule stability and suggest that HDAC6 regulates K394 acetylation during synaptic morphogenesis (Saunders, 2022).
Drosophila Cbp53E regulates axon growth at the meuromuscular junction
Calcium is a primary second messenger in all cells that functions in processes ranging from cellular proliferation to synaptic transmission. Proper regulation of calcium is achieved through numerous mechanisms involving channels, sensors, and buffers notably containing one or more EF-hand calcium binding domains. The Drosophila genome encodes only a single 6 EF-hand domain containing protein, Cbp53E, which is likely the prototypic member of a small family of related mammalian proteins that act as calcium buffers and calcium sensors. Like the mammalian homologs, Cbp53E is broadly though discretely expressed throughout the nervous system. Despite the importance of calcium in neuronal function and growth, nothing is known about Cbp53E's function in neuronal development. To address this deficiency, novel null alleles of Drosophila Cbp53E were generated, and neuronal development was examined at the well-characterized larval neuromuscular junction. Loss of Cbp53E resulted in increases in axonal branching at both peptidergic and glutamatergic neuronal terminals. This overgrowth could be completely rescued by expression of exogenous Cbp53E. Overexpression of Cbp53E, however, only affected the growth of peptidergic neuronal processes. These findings indicate that Cbp53E plays a significant role in neuronal growth and suggest that it may function in both local synaptic and global cellular mechanisms (Hagel, 2015).