The Interactive Fly
Genes involved in tissue and organ development
The organisational principles of locomotor networks are less well understood than those of many sensory systems, where in-growing axon terminals form a central map of peripheral characteristics. Using the neuromuscular system of the Drosophila embryo as a model and retrograde tracing and genetic methods, principles underlying the organisation of the motor system have been uncovered. Dendritic arbors of motor neurons, rather than their cell bodies, are partitioned into domains to form a myotopic map, which represents centrally the distribution of body wall muscles peripherally. While muscles are segmental, the myotopic map is parasegmental in organisation. It forms by an active process of dendritic growth independent of the presence of target muscles, proper differentiation of glial cells, or (in its initial partitioning) competitive interactions between adjacent dendritic domains. The arrangement of motor neuron dendrites into a myotopic map represents a first layer of organisation in the motor system. This is likely to be mirrored, at least in part, by endings of higher-order neurons from central pattern-generating circuits, which converge onto the motor neuron dendrites. These findings will greatly simplify the task of understanding how a locomotor system is assembled. These results suggest that the cues that organise the myotopic map may be laid down early in development as the embryo subdivides into parasegmental units (Landgraf, 2003).
The analysis began by correlating the positions of motor neuron dendrites with the distribution of their muscle targets in the periphery. Motor neurons were retrogradely labelled in a pairwise fashion and the positions of their dendritic arbors were mapped. Because of an interest in the mechanisms that underlie the assembly of the motor system, focus was placed on stages when each motor neuron first establishes a characteristic domain of arborisation within the neuropile (early stage 17, 15h after egg-laying [AEL]) (Landgraf, 2003).
Motor axons project into the muscle field via two main nerves, the intersegmental (ISN) and the segmental nerve (SN). The transverse nerve (TN) runs along the segment border and has few motor axons. Choice of nerve root is one of several features that divide the motor neurons into two principal sets, the ISN and SN. (1) The cell bodies of SN motor neurons are located in the same segment as the muscles that they innervate, whereas ISN motor neuron somata are located in the segment next anterior (with the exception of the RP2 and two neuromodulatory efferent ventral unpaired median [VUM] neurons. (2) ISN motor neurons innervate internal muscles, which span a segment from anterior to posterior, whereas SN (and the TN) motor neurons innervate external muscles. External muscles are distinct from the internal set in several respects: (1) they are generally transverse; (2) unlike internal muscles, they require wingless (wg) signalling for their specification; (3) external (but not internal) muscles and their innervating motor neurons express the cell adhesion molecule (CAM) Connectin, with the single exception of muscle ventral transverse 1 (VT1) (Landgraf, 2003 and references therein).
In addition, ISN and SN motor neurons elaborate their dendrites in distinct regions of the neuropile. Dendrites of ISN motor neurons occupy a domain extending posteriorly from the posterior part of one neuromere into the anterior part of the next. SN motor neuron dendrites occupy a domain that lies between the domains of ISN motor neuron arbors (Landgraf, 2003).
Thus, the organisation of the body wall muscles into internal and external sets is reflected centrally in patterns of motor neuron arborisations. The innervating motor neurons project their axons through different nerves and elaborate their dendritic fields in distinct regions of the neuropile. Although dendritic arbors become progressively more elaborate and extensive over developmental time, their separate domains remain clearly recognisable and appear to be maintained at least until the motor system is fully functional (18 h AEL) (Landgraf, 2003).
Having established that there is a central representation of the muscle field, the organisation of the motor neuron dendrites was analyzed in greater detail. (1) The set of external muscles and their innervating (SN) motor neurons were examined. Muscles of similar anteroposterior positions, such as the ventral acute muscle (VA3) and the segment border muscle (SBM), are innervated by motor neurons whose dendritic arbors lie in a common region of the neuropile. Conversely, motor neurons supplying the anterior (lateral transverse 1-2 [LT1-LT2]) versus the posterior (SBM) muscles have dendritic arbors that are correspondingly separated in the anteroposterior axis of the CNS (Landgraf, 2003).
To put the idea of a regular map to the test, focus was placed on an unusual external motor neuron-muscle pair. Muscle VT1 is innervated by a TN rather than an SN motor neuron. However, VT1 lies at the same place in the anteroposterior axis as the SBM, although VT1 is ventral and the SBM more dorsal. The VT1 motor neuron dendritic field is found to overlaps with that of the SBM motor neuron. For the external set, it is concluded that differences in target muscle location in the anteroposterior axis are mapped centrally as regular differences in dendritic position, but dorsoventral distinctions are not (Landgraf, 2003).
It was next asked whether there is a similarly regular representation of the internal muscles in the developing CNS. While most external muscles are transverse and have unique anteroposterior locations, the internal muscles span the width of a segment so that positional distinctions between them are solely in the dorsoventral axis. It was found that the set of internal muscles is represented centrally by three dendritic domains. Motor neurons innervating ventral internal muscles elaborate their dendritic arbors in the anterior half of the ISN dendritic domain. Motor neurons with dorsolateral internal muscle targets (lateral longitudinal [LL] 1, dorsal acute [DA] 3, dorsal oblique 3-5 [DO3-DO5]) put their arbors into the posterior part of the ISN dendritic domain. Finally, dorsal muscles are represented by a motor neuron dendritic domain that lies between those representing ventral (anterior) and dorsolateral (posterior) internal muscle groups. Thus, the internal muscles are represented in the neuropile by three domains of dendritic arborisation that reflect their different dorsoventral locations in the periphery. Once again, it is concluded that there is a regular mapping of muscle position in the neuropile: in this case, it is positions in the dorsoventral axis peripherally that are represented centrally as differences in the anteroposterior locations of dendrites (Landgraf, 2003).
To test the idea that dendritic arbor positions relate to the distribution of muscles, an atypical motor neuron-muscle pair was examined. The RP2 motor neuron is reported to innervate dorsal muscle DA2, yet its dendrites span the domains that represent both dorsal and dorsolateral internal muscles. However, on careful analysis it was found that DA2 is, in fact, specifically innervated by a U neuron whose dendrites lie in the dorsal internal domain, whereas the RP2 axon forms endings generally on all dorsolateral and dorsal muscles by 19 h AEL. These seem to correspond to the type 1s boutons found in late larvae. Thus, the RP2 neuron puts its dendrites into a region of the neuropile that does indeed represent its targets, namely the dorsolateral and dorsal internal muscles (Landgraf, 2003).
Like the muscle field itself, the map of motor neuron dendrites is metamerically repeated. However, the boundaries of these two units are out of register with one another, since the dendrites of the motor neurons innervating internal muscles lie in the next anterior neuromere. The anterior border of the dendritic map, as defined by the extent of these anterior dendrites, coincides with the anterior margin of engrailed (en) expression. Thus, while the muscles are segmental in their organisation, the domains occupied by the dendrites of their innervating motor neurons are parasegmental (Landgraf, 2003).
To test whether genes that implement the parasegmental pattern in the epidermis are also required for the formation of the parasegmental organisation of the neuromuscular system, the formation of SN and ISN dendritic fields was studied in embryos singly mutant for the following segment polarity genes: en/invected (Df(enE)), wg (wgCX4), naked (nkd2), patched (ptc9), hedgehog (hh21), and gooseberry (Df2R(gsb)). Every one of the six different mutants that were analysed has partially aberrant patterns of neuroblasts (NBs). Nevertheless, SN and ISN motor neurons still form and can be identified by their characteristic axonal projections into the periphery. In addition, it was found that the fundamental separation between SN and ISN dendritic domains is present despite often severe perturbations in CNS structure. For example, in gsb mutant embryos, both nerve roots are frequently fused so that the SN and ISN share a common CNS exit point. Nevertheless, SN and ISN axons as well as their dendritic fields do not intermingle but remain separate. These results suggest that the subdivision of the neuropile into the principal ISN and SN dendritic domains is a robust feature of the system, which appears to be specified early in development, since the embryo subdivides into parasegmental units (Landgraf, 2003).
It was next asked what mechanisms underlie the formation of the myotopic map. Because ISN and SN motor neurons lie at different positions in the CNS and their axons grow out into the muscle field through different nerves, it is reasonable to suppose that at least the major subdivision of dendritic arborisations into internal and external domains could be a byproduct of the locations at which the motor neurons are generated and the paths taken by their growing axons. This passive mapping' explanation can be excluded by considering a single motor neuron-muscle pair, namely dorsal transverse 1 (DT1) and its innervating motor neuron. DT1 is an external muscle (by position, orientation, wg dependence, and Connectin expression), yet its motor neuron is clustered with the internal muscle innervating set and its axon (uniquely for the external muscles) grows out through the ISN. Despite its packing within the internal motor neuron' set, the DT1 motor neuron makes a long posterior projection through the internal muscle domain of the myotopic map to reach the external domain, where it arborises appropriately, reflecting the orientation and external nature of its target muscle. In contrast, motor neurons derived from the same NB as DT1 innervate neighboring internal muscles DO3-DO5 and put their dendrites in a more anterior region characteristic of the dorsolateral muscles. These findings strongly suggest that the mapping of the muscle field within the CNS is an active process of growth and arborisation that partitions dendrites into subdomains of the neuropile that are appropriate to their function, rather than a passive subdivision of available space by position of origin or axon trajectory (Landgraf, 2003).
Since dendritic arbors form after motor axons have reached their targets, the muscles could be instrumental in dictating the organisation of the central map. To test this idea, the UAS/GAL4 system was used to misexpress an activated form of Notch (Kidd et al. 1998) in the developing mesoderm, suppressing the formation of muscle founder cells while leaving other tissues intact. In such muscleless embryos, the main nerve trunks, SN and ISN, still form and project into the periphery. Retrograde labellings of these nerves show that SN and ISN motor neurons form relatively normal dendritic arbors that consistently conform to the characteristic separation of SN and ISN dendrites. Thus, the neuropile is partitioned into distinct fields of dendritic arborisation independently of the muscles. It is concluded that the mapping process is likely to be an autonomous property of the motor neurons and their neighboring cells (Landgraf, 2003).
It was next asked whether motor neuron dendritic fields could be patterned by the substrates on which they grow. In the Drosophila ventral nerve cord (VNC), motor neuron dendrites form in the dorsal-most region of the neuropile, sandwiched between longitudinal glia above and the underlying scaffold of axons. Glial cells can act as substrates for supporting and guiding axonal growth. To test whether they might also be required for the growth and spatial patterning of dendritic fields, dendritic arbors were analysed in glial cells missing (gcm) mutant embryos, which are defective in glial cell differentiation. Although the structure of the nervous system is disrupted in gcm mutant embryos and the dendritic arbors are abnormal, they continue to form in their characteristic locations and the fundamental distinction between the ISN and SN motor neuron dendritic fields is maintained. Remarkably, even the long posterior dendritic projection of the DT1 motor neuron forms and reaches its target region, the SN external muscle dendritic domain. These results suggest that the patterning of the neuropile into distinct motor neuron dendritic domains is a process that appears to be intrinsic to the motor neurons and their neighboring neurons, but does not require proper glial cell differentiation (Landgraf, 2003).
One likely explanation for the division of dendrites into separate domains is that there is a process of mutual exclusion between the arborisations of neighboring cells. Such a process of dendritic tiling' has so far only been documented between particular classes of sensory neurons, but could also occur in the motor system. The idea of tiling was tested by considering two groups of motor neurons whose axons have a common trajectory, but whose dendritic fields form in adjacent territories. The DO3-DO5 and DT1 motor neurons project their dendrites posteriorly, and at their most-anterior point, these dendrites meet the axons and dendrites of the anterior corner cell (aCC) and U/CQ neurons. To show whether the aCC and U/CQ axons and/or dendrites inhibit the growth of DO3-DO5 and DT1 dendrites anteriorly, these neurons (as well as RP2 and the posterior corner cell [pCC] interneuron) were selectively ablated. Using anti-Even-skipped (Eve) staining as a marker for aCC, RP2, and U/CQs (there are an additional two medially located eve-expressing interneurons, pCC and friend of pCC [fpCC], it was found that these neurons can be selectively ablated before they form dendrites (at approximately 11 h AEL): on average, by 10.5 h AEL all but 0.6 and by 12 h AEL all but 0.06 of the seven medially located eve-expressing neurons have been ablated per half-neuromere. In no instance was a concomitant anterior expansion of the DO3-DO5 and DT1 motor neuron dendrites into the regions vacated by the aCC and U/CQ dendrites observed. It is concluded that, at least in this instance, the initial dendritic territory of one set of motor neurons (DO3-DO5 and DT1) is not defined by a process of tiling, in which they are excluded by neighboring (aCC and U/CQ) dendritic arbors. However, it is possible that the elaboration of motor neuron dendritic arbors during later developmental stages may involve interactions between neighboring dendritic territories, activity-dependent processes, or both (Landgraf, 2003).
Thus, in summary, these results suggest that the mechanisms that subdivide the neuropile into distinct dendritc domains are very robust and refractory to perturbations. They further suggest that the cues that organise the map may be laid down early in development as the embryo subdivides into parasegmental units (Landgraf, 2003).
The patterning of the motor neuron dendritic arbors in the Drosophila embryo represents a first layer of organisation in the motor system. This is likely in part to be mirrored by the endings of higher-order neurons of central pattern generating circuits, which converge onto the myotopic map. While motor neuron cell body positions may, as has been proposed for vertebrate systems, relate to the ontogeny of target muscles, the operation of mature muscles is reflected by the allegiance of corresponding motor neuron dendrites to a particular territory in the neuropile. Thus, changes in muscle operation could be accommodated by a change of allegiance of the appropriate motor neuron dendrites from one domain to another (e.g., the DT1 motor neuron-muscle pair) without the need for rewiring the underlying higher-order circuitry. Such a model resolves the apparent discrepancy between the distributions of motor neuron cell bodies centrally and target muscles in the periphery. It also implies a considerable degree of flexibility, particularly at the level of motor output, yet suggests that elements of the underlying motor circuitry may have been highly conserved (Landgraf, 2003).
Glial cells play important roles in the developing brain during axon fasciculation, growth cone guidance, and neuron survival. In the Drosophila brain, three main classes of glia have been identified including surface, cortex, and neuropile glia. While surface glia ensheaths the brain and is involved in the formation of the blood-brain-barrier and the control of neuroblast proliferation, the range of functions for cortex and neuropile glia is less well understood. This study used the nirvana2-GAL4 driver to visualize the association of cortex and neuropile glia with axon tracts formed by different brain lineages and to selectively eliminate these glial populations via induced apoptosis. The larval central brain consists of approximately 100 lineages. Each lineage forms a cohesive axon bundle, the secondary axon tract (SAT). While entering and traversing the brain neuropile, SATs interact in a characteristic way with glial cells. Some SATs are completely invested with glial processes; others show no particular association with glia, and most fall somewhere in between these extremes. The results demonstrate that the elimination of glia results in abnormalities in SAT fasciculation and trajectory. The most prevalent phenotype is truncation or misguidance of axon tracts, or abnormal fasciculation of tracts that normally form separate pathways. Importantly, the degree of glial association with a given lineage is positively correlated with the severity of the phenotype resulting from glial ablation. Previous studies have focused on the embryonic nerve cord or adult-specific compartments to establish the role of glia. This study provides, for the first time, an analysis of glial function in the brain during axon formation and growth in larval development (Spindler, 2009).
Secondary neurons, which are born during the larval period, form SATs that have to extend over relatively long distances, finding their way amidst a complex array of (primary) axons, dendrites, and glia. The association of glia and SATs varies for different lineages. SATs either (1) remained wrapped within the neuropile or joined other tracts that were then ensheathed as a larger tract system, (2) encountered strands of glial condensations, or (3) had no association with glial sheaths. The association between individual SATs and glia was highly invariant. Thus, if SAT A joined SAT B to form a larger tract system that became wrapped by glia, the same densities of glia could be observed in other brains for the corresponding SATs (Spindler, 2009).
To address the role of glia during SAT fasciculation, growth, and guidance, cortex and neuropile glia, the two glial types in contact with growing SATs, were selectively eliminate. Expression of the pro-apoptotic proteins Hid and Rpr were effective in inducing apoptosis of most cortex and neuropile glia by the early or mid larval stage. It can be assumed that the primary axon tract formation is not disrupted, given that expression of the
nrv2-GAL4 driver line does not set in prior to stage 12, primary axon tract (PAT) patterning is complete before glia invade the neuropile, and the first signs of apoptosis appear after hatching. In addition, the surface glia remain intact, providing general ensheathment around the brain, a functional blood–brain barrier, and potential signaling molecules used to control neuroblast proliferation (Spindler, 2009).
Upon the elimination of glia, frequent abnormalities are seen in the pattern of SATs. Importantly, the strength of an SAT phenotype appears to correlate with the degree of glial association of that SAT in a wild-type brain. Of all SATs analyzed, the mushroom body (associated with a complete glial sheath) exhibits the most severe defects, including complete SAT misguidance and aberrant fasciculation of neurites from adjacent SATs. In contrast, SATs that were less endowed with glia in the wild type typically had a normal projection pattern (Spindler, 2009).
This study does not differentiate between a role for glia in producing chemo-attractant signals or acting as a physical scaffold for guidance. In previous studies, ectopic expression of dominant-negative Drosophila E-cadherin in either cortex/neuropile glia or SATs themselves results in non-radial trajectories of SATs into the neuropile. This suggests a requirement for SAT-glia adhesion as secondary axons project toward the cortex-neuropile boundary. However, the direction of neuroblast division is also disrupted with DE-cadherin knock-down, thus aberrant SAT trajectories
may be a secondary effect of abnormal cell body layering within the cortex. In support of a signaling role for glia, the midline guidance defect of the CP1 SAT (anteromedially, crossing the peduncle and entering the diagonal commissure) is reminiscent of the robo-slit phenotypes in the
Drosophila ventral nerve cord. Whether robo/slit signaling is also used between SATs and glia in the brain is a question that warrants further investigation (Spindler, 2009).
The situation that SATs in part require glia for pathway guidance in the neuropile is different than the embryonic brain in which pioneer axons and PATs are formed in the absence of extensive glial processes. Why are SATs different? One can imagine the scenario in which the PATs generate the neuropile de novo, forcing cell body movement outwards as the central neuropile grows. By first instar, a full neuropile is established, and SATs must guide through a dense maze of neurites, glia, and trachea. It therefore
appears that: (1) PATs establish the initial connectivity of the brain; (2) glia grow in around this initial scaffolding, and finally (3) SATs use both the PAT scaffolding and the glial boundaries for guidance into and around the neuropile (Spindler, 2009).
Insects have long been used to evaluate glial-neuronal interactions from embryonic to adult stages. An important focus of these studies was whether or not glia or axon tracts appear first, and in how far axonal pathfinding is disrupted if glia is ablated. In this regard, clear developmental differences have been found between distinct regions of the nervous system, as well as between insect species for homologous nervous system domains (Spindler, 2009).
In the Drosophila ventral nerve cord, two subpopulations of neuropile glia were studied in the context of axonal pathfinding: midline glia and longitudinal glia. Both types of glial cells appear around the same stage when pioneer neurons extend their axons. The proper number and positioning of midline glia is clearly required for the formation of commissural axon tracts. The loss of longitudinal glia (by ablation and in embryos mutant for the
gcm gene) primarily affects the defasciculation and fasciculation events of longitudinal pioneer tracts, subsequently affecting the follower neuron trajectories. Note that gcm is required for all classes of glia, including surface glia; thus, in very late gcm mutant embryos, severe disruptions of the entire neuropile result from the fact that, with the onset of embryonic movement, the CNS lacking surface glia is literally 'shredded to pieces' (Spindler, 2009).
In the embryonic Drosophila peripheral nervous system, ablation of the peripheral glia (i.e. exit glia) via targeted overexpression of the cell death genes grim and ced-3 lead to aberrant pathfinding of both motor and sensory axons as they exited the CNS; although the motor neurons eventually overcame the absence of peripheral glia finding correct muscle targets, suggesting a limited role for the peripheral glia in the initial trajectory of motor neurons. In grasshopper, ablation of the cell-segment boundary glial guidepost cell lead to more severely aberrant axon trajectories. Ablation of glia surrounding the antennal lobe of adult Manduca generates olfactory axon de-fasciculation and misguidance. Finally, guidance phenotypes have also been observed with disruption of the Drosophila lamina glia; R1-R6 photoreceptor axons show aberrant guidance past the lamina into the medulla of the optic lobe (Spindler, 2009 and references therein).
An important aspect of the developmental role of glia added by this study is the focus on the correlation between closeness of axon tract/glia association, and axon tract abnormality in the absence of glia. In other words: the nervous system is formed by a large number of fascicles, and these fascicles vary in their degree of glial wrapping. It would be misleading when carrying out a genetic study to only focus on a single fascicle (or small subset of fascicles), and extrapolate from the phenotype observed for this fascicle onto the brain as a whole. In this analysis, SATs of several lineages, notably those that in normal brains have little glia covering them in the neuropile, show few abnormalities in glia-less brains. By contrast, other lineages were affected in the majority of cases, and typically, these lineages also were the ones whose SATs were associated more closely with glia (Spindler, 2009).
Neuropile compartments are formed by terminal branches of axons and dendrites and their synapses. For example, the antennal lobe of insects consists of the axonal terminals of sensory neurons located in the antenna, and dendritic terminals of antennal projection neurons and local interneurons located in the deuterocerebrum, aside from a relatively small number of other modulatory neurons. Sensory neurons expressing the same olfactory receptor all converge onto the dendrites of a small number of projection neurons
to form an olfactory glomerulus. In many insects, notably Manduca, olfactory glomeruli are individually compartmentalized by neuropile glia, and it has been shown that glia plays a prominent role in establishing the glomeruli organization. According to the prevailing view, glomeruli are initially ordered by the specialized endings of sensory terminals into protoglomeruli; however, glial processes soon invade the space in between protoglomeruli and restrict the arborization of receptor axons. Therefore, in Manduca antennal lobes, glia is required for early maintenance of the glomerular map (Spindler, 2009 and references therein).
A recent analysis of the time course of glial development in the Drosophila antennal lobe suggested that in this species, glia plays an even lesser role, since glomeruli are only incompletely, and at a late time point, wrapped by glia. While glia were never ablated in those studies, later work found that elimination of Neuroglian from midline glia resulted in an inability of ORN axons to cross through the antennal commissural tract to the contralateral lobe. In this study, most adult brains lacking cortex and neuropile glia still form antennal lobes with glomeruli, however in some samples the glomeruli are poorly defined, and cannot be identified by their position and shape in the antennal lobe. The variability in phenotype penetrance likely stems from larvae containing the most glia surviving to eclosion; therefore, a relatively normal looking adult brain could stem from a weak glial apoptosis early in development. Whether the antennal lobe
disorganization is a secondary defect due to a lack of definition normally provided by the presence of glia, a result of SATs that normally contribute to the AL misguiding or truncating early, or a maintenance defect in which axons begin inappropriately intermingling among glomeruli is not clear. Perhaps the Drosophila antennal lobe glia is required for glomeruli maintenance even after glomeruli organization is established, and future studies will hopefully address this possibility (Spindler, 2009).
In the post-embryonic midline of Drosophila, ablation of the transient interhemispheric fibrous ring (TIFR), a transient population of midline glia, by ectopic expression of the pro-death gene hid, generates defects in the adult central complex. What is unclear, however, is the cellular event that is effected to cause the defects. This study suggests that morphological abnormalities in adult compartments from glial manipulation are due to the misguidance of larval SATs to the correct neuropile compartment in the brain, affecting the formation of adult neuropile structures. This study presents evidence that glia is an important mediator of axon guidance in the Drosophila larval brain; the mechanism for glia-neuron communication during this process is an exciting area for future investigation (Spindler, 2009).
Axons damaged by acute injury, toxic insults, or during neurodegenerative diseases undergo Wallerian or Wallerian-like degeneration, which is an active and orderly cellular process, but the underlying mechanisms are poorly understood. Drosophila has been proven to be a successful system for modeling human neurodegenerative diseases. This study established a novel in vivo model of axon injury using the adult fly wing. The wing nerve highlighted by fluorescent protein markers can be directly visualized in living animals and be precisely severed by a simple wing cut, making it highly suitable for large-scale screening. Using this model, an axonal protective function of WldS and nicotinamide mononucleotide adenylyltransferase (Nmnat) was confirmed. It was further revealed that knockdown of endogenous Nmnat triggered spontaneous, dying-back axon degeneration in vivo. Intriguingly, axonal mitochondria were rapidly depleted upon axotomy or downregulation of Nmnat. The injury-induced mitochondrial loss was dramatically suppressed by upregulation of Nmnat, which also protected severed axons from degeneration. However, when mitochondria were genetically eliminated from axons, upregulation of Nmnat was no longer effective to suppress axon degeneration. Together, these findings demonstrate an essential role of endogenous Nmnat in maintaining axonal integrity that may rely on and function by stabilizing mitochondria (Fang, 2012).
This study presents a novel in vivo model for axon injury and degeneration based on the adult fly wing. Using this model, the following was uncovered: (1) endogenous dNmnat is required for axonal integrity, (2) axonal mitochondria are depleted rapidly upon axotomy or downregulation of dNmnat, (3) upregulation of dNmnat preserves mitochondria in injured axons and delays Wallerian degeneration, and (4) removal of mitochondria from axons abolishes the protective effect of WldS and Nmnat. The levels of mNmnat2 rapidly decline in mammalian neurite culture upon injury. Drosophila has only one gene encoding Nmnat, and axon degeneration was observed as early as 18 hr after axotomy, suggesting a rapid turnover of dNmnat. Hence, reduction of Nmnat levels, either by rapid turnover of Nmnat upon axotomy or genetic knockdown of dNmnat, may render mitochondria unstable and/or dysfunctional, thus triggering axon degeneration. The self-destructive mechanisms of axon degeneration in injury and loss of endogenous Nmnat appear to converge at axonal mitochondria. This may underlie the morphological similarity between Wallerian degeneration and spontaneous axon degeneration in dying-back diseases. As such, endogenous Nmnat and axonal mitochondria may be key to identifying additional downstream events and therefore providing exciting new targets for therapeutic interventions of both acute neural injury and chronic axonal disorders (Fang, 2012).
WldS (slow Wallerian degeneration) is a remarkable protein that can suppress Wallerian degeneration of axons and synapses, but how it exerts this effect remains unclear. Using Drosophila and mouse models, this study identified mitochondria as a key site of action for WldS neuroprotective function. Targeting the NAD(+) biosynthetic enzyme Nmnat to mitochondria was sufficient to fully phenocopy WldS, and WldS was specifically localized to mitochondria in synaptic preparations from mouse brain. Axotomy of live wild-type axons induced a dramatic spike in axoplasmic Ca(2+) and termination of mitochondrial movement-WldS potently suppressed both of these events. Surprisingly, WldS also promoted increased basal mitochondrial motility in axons before injury, and genetically suppressing mitochondrial motility in vivo dramatically reduced the protective effect of WldS. Intriguingly, purified mitochondria from WldS mice exhibited enhanced Ca(2+) buffering capacity. It is proposed that the enhanced Ca(2+) buffering capacity of WldS+ mitochondria leads to increased mitochondrial motility, suppression of axotomy-induced Ca(2+) elevation in axons, and thereby suppression of Wallerian degeneration (Avery, 2012).
The mechanistic action of WldS has remained controversial, but recent work has established a nonnuclear role for WldS after injury. This study shows that WldS is localized to mitochondria in vivo. It is important to note that protein localization studies with WldS must be interpreted cautiously -- the primarily nuclear localization of WldS suggested a nuclear role for WldS and initially misled the field. However, this study also found that WldS increases mitochondrial Ca2+ buffering capacity and results in maintained mitochondrial motility after axotomy. Taken together, these data argue strongly that the mitochondrial compartment is a key site of action for WldS in vivo (Avery, 2012).
This study has shown that axonal injury in live Drosophila preparations leads to a dramatic and transient rise in axonal Ca2+. Increased axonal Ca2+ has been observed in mammals after acute nerve crush and entry of extracellular Ca2+ is necessary and sufficient for Wallerian degeneration. Impressively, WldS expression resulted in a striking suppression of this axotomy-induced rise in axonal Ca2+. The most plausible explanation for this enhanced buffering is that increased ATP and energy production observed in WldS+ mitochondria -- presumably via increased mitochondrial NAD+ production, though essential roles cannot be formally excludes for other substrates of Nmnat -- is linked to increased mitochondrial membrane potential (ΔΨm), and thereby increased Ca2+ entry through the ΔΨm-regulated mitochondrial Ca2+ uniporter. This model is supported by the observation that WldS-expressing mitochondria isolated from mouse brain exhibit an enhanced ability to maintain their membrane potential and avoid PTP formation in the face of increasing extramitochondrial Ca2+. In the future, it will be important to confirm that such changes are also observed in Drosophila axonal mitochondrial physiology in vivo in WldS-expressing neurons (Avery, 2012).
Axonal Ca2+ spikes could result solely from entry of extracellular Ca2+ into the axon after injury. This would be consistent with the observation that blocking Ca2+ channels inhibits Wallerian degeneration. Mitochondria are a well-established sink for Ca2+ in axons and this study shows that WldS+ mitochondria exhibit enhanced Ca2+ buffering capacity and resistance to Ca2+-induced formation of the permeability transition pore (PTP). Indeed PTP formation appears to be a key final execution step in Wallerian degeneration. A model is therefore favored whereby extracellular Ca2+ enters the axon after axotomy and normally acts as a switch to activate Wallerian degeneration. In WldS axons, this Ca2+ is instead rapidly buffered by mitochondria, thereby blocking induction of axonal destruction. Consistent with this model, uncoupling mitochondria, which suppresses mitochondrial Ca2+ uptake, completely abrogates the protective effect of WldS in vitro (Avery, 2012).
WldS-expressing neurons exhibit a roughly 2-fold increase in the number of motile versus stationary mitochondria compared to WT controls, which could result from changes in mitochondrial Ca2+ buffering. Notably, genetic suppression of enhanced mitochondrial flux using mutations in miro also resulted in a remarkable suppression of WldS-mediated axonal protection in vivo. However, because this suppression was only partial, additional factors beyond increases in mitochondrial motility must also contribute to WldS-mediated axonal protection. For example, axonal energy supplies are likely closely intertwined with mitochondrial transport and bioenergetics. WldS+ mitochondria are known to exhibit an enhanced ability to generate ATP. This change in bioenergetics, coupled with increased mitochondrial motility in WldS+ axons, might enhance distribution of ATP or other mitochondrially-derived metabolites. At the same time, enhanced mitochondrial motility could also speed the removal of metabolic byproducts normally processed by mitochondria. Similarly, increased mitochondrial motility in axons could further enhance mitochondrial Ca2+ buffering in WldS+ axons because motile mitochondria would be predicted to traverse more 'axonal space' and perhaps be exposed to more Ca2+ than stationary mitochondria. Together, these could have the combined effect of increasing energy delivery, removing harmful byproducts, and increased buffering of Ca2+, a signal that can potently activate axonal degeneration (Avery, 2012).
A role for mitochondria in the WldS neuroprotective mechanism is intriguing because defects in mitochondria respiration and dynamics are emerging as critical underlying factors in a number of neurological disorders. For example, in mouse models of ALS (SOD1 transgenics), anterograde and retrograde mitochondrial transport is reduced, altered mitochondrial trafficking has been observed in models of Alzheimer's disease, and mutant, but not WT Huntington, protein blocks mitochondrial movement in cortical neurons. However, in the majority of models, whether these mitochondrial alterations are a cause or consequence of disease remains an open question. The current study shows, reciprocally, that enhanced mitochondrial flux is associated with and is required for maximal axon protection by WldS (Avery, 2012).
In vitro studies conducted in Aplysia and chick sensory neurons indicate that in addition to microtubule assembly, long microtubules in the C-domain of the growth cone move forward as a coherent bundle during axonal elongation. Nonetheless, whether this mode of microtubule translocation contributes to growth cone motility in vivo is unknown. This question was addressed in Drosophila. Using docked mitochondria as fiduciary markers for the translocation of long microtubules, motion along the axon was examined to test if the pattern of axonal elongation is conserved between Drosophila and other species in vitro. When Drosophila neurons were cultured on Drosophila extracellular matrix proteins collected from the Drosophila Kc167 cell line, docked mitochondria moved in a pattern indicative of bulk microtubule translocation, similar to that observed in chick sensory neurons grown on laminin. To investigate whether the C-domain is stationary or advances in vivo, the movement of mitochondria was tracked during elongation of the aCC motor neuron in stage 16 Drosophila embryos. Docked mitochondria were found to move forward along the axon shaft and in the growth cone C-domain. This work confirms that the physical mechanism of growth cone advance is similar between Drosophila and vertebrate neurons and suggests forward translocation of the microtubule meshwork in the axon underlies the advance of the growth cone C-domain in vivo. These results highlight the need for incorporating en masse microtubule translocation, in addition to assembly, into models of axonal elongation (Roossien, 2013).
The Rac-Cofilin pathway is essential for cytoskeletal remodeling to control axonal development. Rac signals through the canonical Rac-Pak-LIMK pathway to suppress Cofilin-dependent axonal growth and through a Pak-independent non-canonical pathway to promote outgrowth. Whether this non-canonical pathway converges to promote Cofilin-dependent F-actin reorganization in axonal growth remains elusive. This study demonstrates that Sickie, a homolog of the human microtubule-associated protein neuron navigator 2, cell-autonomously regulates axonal growth of Drosophila mushroom body (MB) neurons via the non-canonical pathway. Sickie was prominently expressed in the newborn F-actin-rich axons of MB neurons. A sickie mutant exhibited axonal growth defects, and its phenotypes were rescued by exogenous expression of Sickie. Phenotypic similarities and genetic interactions were observed among sickie and Rac-Cofilin signaling components. Using the MARCM technique, distinct F-actin and phospho-Cofilin patterns were detected in developing axons mutant for sickie and Rac-Cofilin signaling regulators. The upregulation of Cofilin function alleviated the axonal defect of the sickie mutant. Epistasis analyses revealed that Sickie suppresses the LIMK overexpression phenotype and is required for Pak-independent Rac1 and Slingshot phosphatase to counteract LIMK. It is proposed that Sickie regulates F-actin-mediated axonal growth via the non-canonical Rac-Cofilin pathway in a Slingshot-dependent manner (Abe, 2014).
During brain development, neurons undergo multiple morphological changes to form an elaborate neural network. The Drosophila mushroom body (MB), which forms bilaterally symmetric and dorsomedially bifurcated axonal lobe structures in the central brain, has been well studied as a model of neuronal development. Among various regulators of neuronal morphogenesis, ADF/Cofilin and Rac GTPase (Rac) are key molecules in controlling cytoskeletal remodeling in axonal development. Cofilin [Twinstar (Tsr) in Drosophila] plays an essential role as a regulator of axonal growth by severing and depolymerizing F-actin. Because Cofilin is activated by dephosphorylation by the Slingshot (Ssh) phosphatase and is inactivated by phosphorylation by LIMK, the loss of Ssh or excessive activation of LIMK results in an axonal growth defect. In Drosophila, Rac has been proposed to act as a bidirectional switch for signaling cascades. One signaling event is the canonical Rac-Pak-LIMK pathway to suppress Cofilin-dependent axonal growth. The overexpression of Pak, a downstream effector of Rac, induces axonal growth defects similar to those observed with LIMK overexpression. In addition, introducing one mutant copy of Rac or Pak suppresses the axonal defect induced by LIMK overexpression. Another signaling event is the Pak-independent non-canonical pathway to positively regulate axonal growth. Rac mutant animals show multiple MB axonal defects, but the axonal growth defect is alleviated by the exogenous expression of Rac1Y40C, which lacks the ability to activate Pak but does not affect lamellipodia formation. Furthermore, Rac1Y40C overexpression remarkably suppressed the LIMK overexpression phenotype (Abe, 2014).
Although several pieces of evidence have suggested the importance of the non-canonical pathway and predicted the existence of its mediator, whether this pathway finally converges upon the downstream Cofilin pathway and subsequent F-actin reorganization remains unclear. Moreover, many biochemical studies have assessed the regulation of Cofilin function and F-actin states using in vitro systems; the endogenous changes in F-actin and Cofilin phosphorylation appear not to have been analyzed simultaneously with an internal control in developing brain. To address these issues, a novel factor was sought that interacts with Rac-Cofilin signaling components and positively regulates MB axonal growth. It was observed that Sickie, which has a calponin homology (CH) actin-binding domain and shares structural similarities with the human microtubule-associated protein (MAP) neuron navigator 2 (NAV2), showed prominent expression in F-actin-rich newborn MB axons and genetically interacted with Rac-Cofilin signaling regulators. Although Sickie was originally identified by genome-wide RNAi screening in Drosophila S2 cells and the report proposed the involvement of Sickie in the innate immune response (Foley, 2004), in this report focus was placed on the function of Sickie in the regulation of Cofilin-mediated F-actin remodeling and propose an expanded model of regulatory mechanisms during axonal development (Abe, 2014).
By combining the MARCM technique with epistatic analysis, this study demonstrated that Sickie regulates the axonal growth of Drosophila MB neurons via the non-canonical Rac-Cofilin pathway. The following model is proposed. In wild type, Sickie relays the non-canonical pathway signal to Ssh to facilitate F-actin-mediated axonal growth by counteracting the canonical signal. In a sickie mutant, mediation of the non-canonical pathway is defective, which causes an imbalance in the regulation of Cofilin activity. Because neurons are morphologically polarized and the amount of actin is limited in each cell, the growing axons may efficiently control actin recycling by facilitating F-actin turnover by balancing between the non-canonical and canonical pathways. Consistently, a stronger axonal growth defect was found with increased P-Cofilin in the LIMKWTM6 ssh1-63 and sickieΔ LIMKKD double-mutant animals than in the single mutants ssh1-63, sickieΔ and LIMKKD. Cofilin activity might be decreased in the developing axons of these double mutants by the preponderance of the canonical pathway. If so, these results highlight an essential role of the non-canonical pathway to balance Cofilin activity in axonal growth (Abe, 2014).
Unlike the clear elevation of P-Cofilin levels in the ssh1-63 mutant, constitutive activation of LIMK did not result in a similar increase in P-Cofilin despite F-actin elevation. This apparent paradox might be explained by considering the positive regulation of Ssh by F-actin. The phosphatase activity of SSH-1L is F-actin dependent, and the addition of F-actin dramatically increases its phosphatase activity. In the LIMKKD mutant axons, endogenous Ssh may be activated by a large amount of F-actin and subsequently dephosphorylates Cofilin. Consistently, highly elevated signals of both F-actin and P-Cofilin were detected in the LIMKWTM6 ssh1-63 double-mutant clones. In this mutant, Cofilin activity was severely reduced by high phosphorylation levels due to constitutive LIMK activation and a lack of Ssh phosphatase activity, resulting in the posterior arrest severe axonal defect, similar to the cofilin knockdown mutant. In addition, relatively moderate increases in P-Cofilin signal were detected in the developing axons of the sickieΔ LIMKKD double mutant. These results also support the model that Sickie functions in the same pathway as Ssh to positively regulate Cofilin function by counteracting the canonical Rac-Pak-LIMK pathway. Ssh might be downregulated in the sickie mutant axons due to defects in the mediation of Pak-independent Rac1 function or in the interaction among Ssh and F-actin by the loss of Sickie. The similar increases in the P-Cofilin and F-actin signals and the similar posterior arrest phenotype in the LIMKWTM6 ssh1-63 double-mutant clone and those of the PakMyr mutant clone are also consistent with results of in vitro studies that showed that SSH-1L is inactivated by Pak4. Thus, in the current model, Pak concurrently inactivates Ssh and activates LIMK in axonal growth (Abe, 2014).
Whereas ssh or cofilin mutants are embryonic lethal and their mutant clones display developmental defects in non-neuronal tissues, sickie mutants are not embryonic lethal, and conspicuous phenotypes are found only in the substructures of the central brain, such as MB and EB, implying that more elaborate mechanisms involving Sickie function are required for ensuring their proper development. Given that MB neurons exhibit a densely bundled axonal morphology, the growing MB axons might require Sickie to smoothly extend their neurites within the lobe core region by coordinating the dynamics of actin and microtubules (MTs). Sickie and human neuron navigator proteins (NAVs) have conserved EB1-binding motifs, and Sickie shows a genetic interaction with MT components. Double RNAi of sickie and EB1 or β-tubulin both resulted in synergistic increases in the axonal defects. In addition, a recent cell biological study demonstrated a functional link between Cofilin and MTs. Through its interaction with EB1, Sickie might act as a navigator for the plus-end of MTs to link to the F-actin complex and thereby ensure elaborate neuronal wiring. To further elucidate the signaling mechanism, the relationships with other components of the Ssh-dependent Cofilin pathway need to be studied. Recent studies have revealed that PKD, 14-3-3 protein and Pak4 play key roles in suppressing Ssh function (Abe, 2014).
Finally, preliminary data suggest a post-developmental role for Sickie. Adult stage-specific knockdown of Sickie in MBs impairs olfactory memory. Moreover, recent mammalian studies have suggested the possible involvement of NAVs and Cofilin in neurodegenerative disease. In Alzheimer's disease brains, the NAV3 transcript level is elevated, and Cofilin-actin rod-shaped inclusions, which are formed by the hyperactivation of Cofilin, are enriched. Further studies are required to understand the wide variety of contributions of sickie and the general importance of this evolutionarily conserved gene in brain development and function (Abe, 2014).
Transcription factors establish neural diversity and
wiring specificity; however, how they orchestrate
changes in cell morphology remains poorly understood.
The Drosophila Roundabout (Robo) receptors
regulate connectivity in the CNS, but how their
precise expression domains are established is unknown.
This study shows that the homeodomain transcription
factor Hb9 acts upstream of Robo2 and
Robo3 to regulate axon guidance in the Drosophila
embryo. In ventrally projecting motor neurons, hb9
is required for robo2 expression, and restoring
Robo2 activity in hb9 mutants rescues motor axon
defects. Hb9 requires its conserved repressor
domain and functions in parallel with Nkx6 to regulate
robo2. Moreover, hb9 can regulate the mediolateral
position of axons through robo2 and robo3,
and restoring robo3 expression in hb9 mutants rescues
the lateral position defects of a subset of neurons.
Altogether, these data identify Robo2 and
Robo3 as key effectors of Hb9 in regulating nervous
system development (Santiago, 2014).
Combinations of transcription factors specify the tremendous diversity
of cell types in the nervous system. Many studies have identified requirements
for transcription factors in regulating specific events
in circuit formation as neurons migrate, form dendritic and
axonal extensions, and select their final synaptic targets. In most cases,
the downstream effectors through which transcription factors
control changes in neuronal morphology and connectivity
remain unknown, although several functional relationships
have been demonstrated (Santiago, 2014).
Conserved homeodomain transcription factors regulate motor
neuron development across phyla. Studies in vertebrates and invertebrates
have shown that motor neurons that project to common
target areas often express common sets of transcription
factors, which act instructively to direct motor axon guidance. In mouse and chick, Nkx6.1/
Nkx6.2 and MNR2/Hb9 are required for the specification of spinal
cord motor neurons, and for axon pathfinding and muscle
targeting in specific motor nerves. In Drosophila, Nkx6 and Hb9 are expressed
in embryonic motor neurons that project to ventral or
lateral body wall muscles, and although they are not individually
required for specification, they are essential for the pathfinding of
ventrally projecting motor axons. Axons that project to
dorsal muscles express the homeodomain transcription factor
Even-skipped (Eve), which regulates guidance in part through
the Netrin receptor Unc5. Eve exhibits cross-repressive interactions
with hb9 and nkx6, which function in parallel to repress
eve and promote islet and lim3. Hb9 and Nkx6 act as repressors to
regulate transcription factors in the spinal cord; however, guidance
receptors that act downstream of Hb9 and Nkx6 have not been
characterized. Interestingly, in both flies and vertebrates, Hb9
and Nkx6 are also expressed in a subset of interneurons, and
knockdown experiments in Drosophila have suggested a role
for hb9 in regulating midline crossing (Santiago, 2014).
Roundabout (Robo) receptors regulate midline crossing and
lateral position within the developing CNS of invertebrates and
vertebrates. Two recent studies in mice have also identified a role for Robos
in regulating motor axon guidance in specific motor neuron
populations. The three Drosophila Robo receptors have
diversified in their expression patterns and functions. Robo2 is initially expressed in many ipsilateral
pioneers and also contributes to Slit-mediated repulsion. Subsequently,
robo2 expression is more restricted, and it is required to specify
the medio-lateral position of axons. Robo3 is expressed in a subset of CNS
neurons and also regulates lateral position (Santiago, 2014).
Characterization of the expression domains of the Drosophila
Robos revealed an intriguing pattern, in which Robo1 is expressed
on axons throughout the width of the CNS, Robo3 is
found on axons in intermediate and lateral zones, and Robo2 is
enriched on the most lateral axons. These patterns are transcriptional in
origin, as replacing any robo gene with the coding sequence
of another Robo receptor results in a protein distribution that
matches the endogenous expression of the replaced gene
(Spitzweck, 2010). A phenotypic analysis of these gene-swap alleles revealed
the importance of transcriptional regulation for the diversification
of robo gene function (Spitzweck, 2010). Robo2 and
robo3's roles in regulating lateral position are largely dependent
on their expression patterns, although unique structures within
the Robo2 receptor are also important for its function in lateral
position (Evans, 2010; Spitzweck, 2010). In
the peripheral nervous system, the Atonal transcription factor
regulates robo3 in chordotonal sensory neurons, directing the
position of their axon terminals. In the CNS,
the transcription factors lola and midline contribute to the induction
of robo1. However, how the expression patterns of robo2 and robo3 are established
to direct axons to specific medio-lateral zones within the CNS
remains unknown (Santiago, 2014).
This study identifies a functional relationship between Hb9 and
the Robo2 and Robo3 receptors in multiple contexts. Hb9 acts through Robo2 to regulate motor axon guidance
and can direct the medio-lateral position of axons in the nerve
cord through its effects on robo2 and robo3. Furthermore, hb9
interacts genetically with nkx6 and requires its conserved
repressor domain to regulate robo2. Together, these data establish
a link between transcriptional regulators and cell surface
guidance receptors, providing an example of how upstream factors
act through specific guidance receptors to direct circuit formation (Santiago, 2014).
This study has demonstrated a functional relationship between Hb9
and the Robo2 and Robo3 receptors in multiple contexts in the
Drosophila embryo. In the RP motor neurons, hb9 is required
for robo2 expression, and genetic rescue experiments indicate
that robo2 acts downstream of hb9. Hb9 requires its conserved
repressor domain and acts in parallel with Nkx6 to regulate robo2
and motor axon guidance. Moreover, hb9 contributes to the
endogenous expression patterns of robo2 and robo3 and the
lateral position of a subset of axons in the CNS, and can redirect
axons laterally when overexpressed via upregulation of robo2.
Finally, restoring Robo3 rescues the medial shift of MP1 axons
in hb9 mutants, indicating that hb9 acts through robo3 to regulate
medio-lateral position in a defined subset of neurons (Santiago, 2014).
Hb9 and nkx6 are required for the expression of robo2 in motor
neurons, and rescue experiments suggest that the loss of
robo2 contributes to the phenotype of hb9 mutants. However,
nkx6 mutants and hb9 mutants heterozygous for nkx6 have a
stronger ISNb phenotype than robo2 mutants, implying the existence
of additional downstream targets. One candidate is the
cell adhesion molecule FasIII, which is normally expressed in
the RP motor neurons and appears reduced in nkx6 mutant
embryos. Identifying the constellation of
effectors that function downstream of Hb9 and Nkx6 will be
key to understanding how transcription factors expressed in
specific neurons work together to drive the expression of the
cell surface receptors that regulate axon guidance and target
selection (Santiago, 2014).
Robo2's activity in motor axon guidance appears distinct
from the previously described activities of the Drosophila Robo
receptors. Although Robo1 can replace Robo2's repulsive activity
at the midline (Spitzweck, 2010), Robo2's function in
motor axon guidance is not shared by either Robo1 or Robo3.
Moreover, Robo2's antirepulsive activity at the midline and its
ability to shift axons laterally when overexpressed both map to
Robo2's ectodomain, whereas this study has found that Robo2's activity
in motor axon guidance maps to its cytodomain (Evans, 2010; Spitzweck, 2010). The signaling outputs of Robo2's cytodomain remain unknown, as it lacks the conserved
motifs within Robo1 that engage downstream signaling partners.
How does Robo2 function during motor axon guidance? In mice,
Robo receptors are expressed in spinal motor neurons and prevent
the defasciculation of a subset of motor axons (Jaworski, 2012). Does Drosophila Robo2 regulate
motor axon fasciculation? The levels of adhesion between
ISNb axons and other nerves must be precisely controlled during
the different stages of motor axon growth and target selection,
and several regulators of adhesion are required for ISNb guidance. Furthermore, whereas Slit can be detected on
ventral muscles, it is not visibly enriched in a pattern that suggests
directionality in guiding motor axons, making it difficult to envision how Robo2-mediated repulsive
or attractive signaling might contribute to ISNb pathfinding.
Future work will determine how Robo2's cytodomain mediates
motor axon guidance, whether this activity is Slit dependent,
and whether Robo2 signals attraction, repulsion, or modulates
adhesion in Drosophila motor axons (Santiago, 2014).
Elegant gene-swap experiments revealed the importance of
transcriptional regulation in establishing the different expression
patterns and functions of the Drosophila Robo receptors (Spitzweck, 2010). By analyzing a previously uncharacterized
subset of axon pathways, this study has uncovered a requirement
for Hb9 in regulating lateral position in the CNS. Although Hb9
can act instructively to direct lateral position when overexpressed,
its endogenous expression in a subset of medially projecting
neurons suggests that its ability to shift axons laterally is
context dependent. A complex picture emerges in which multiple
factors act in different groups of neurons to regulate robo2
and robo3. In a subset of interneurons, hb9 is endogenously
required for lateral position through the upregulation of robo3
and likely robo2. In other neurons, such as those that form the
outer FasII tracts, the expression patterns of robo2 and robo3
rely on additional upstream factors. What might be the significance
of a regulatory network in which multiple sets of transcription
factors direct lateral position in different groups of neurons?
One possibility is that hb9-expressing neurons may share specific
functional properties, such as the expression of particular
neurotransmitters or ion channels. Alternatively, hb9 may regulate
other aspects of connectivity. Indeed, Robo receptors
mediate dendritic targeting in the Drosophila CNS, raising the exciting possibility that hb9 regulates both
axonal and dendritic guidance through its effects on guidance receptor expression (Santiago, 2014).
What is the mechanism by which Hb9 regulates the expression
of robo2, robo3, and its other downstream effectors? This study has
found that Hb9 requires its conserved putative repressor domain
and acts in parallel with Nkx6 to regulate robo2 and motor axon
guidance. It has previously been shown that hb9 and nkx6 function
in parallel to regulate several transcription factors. Hb9, nkx6 double mutants
show decreased expression of islet and lim3 and upregulation
of eve and the Nkx2 ortholog vnd. Are Hb9 and Nkx6 regulating robo2 or robo3 through any of their previously identified targets? Hb9 and nkx6 single mutants show no
change in islet, lim3, or vnd expression, arguing that hb9 and nkx6 do not
act solely through these factors to regulate robo2 or robo3.
Eve expression is unaffected in nkx6 mutants, and whereas it is ectopically expressed in two neurons
per hemisegment in hb9 mutants, these do not correspond to RP3 or MP1, the identifiable cells
in which changes can be detected in robo2 and robo3. Therefore, the data do not support the hypothesis that
Hb9 and Nkx6 regulate robo2 or robo3 primarily through their previously identified targets islet, lim3, vnd, or eve (Santiago, 2014).
Gain-of-function experiments in vertebrates suggest that Hb9
and Nkx6 act as repressors to regulate gene expression in the
spinal. The finding that Hb9's Eh domain is required for motor
axon pathfinding and robo2 regulation suggests that Hb9 acts
as a repressor in this context as well, most likely through a previously
unidentified intermediate target. In contrast, the
Eh domain is not required for Hb9's ability to regulate robo3 or
lateral position in hb9GAL4+ neurons that project to intermediate
zones of the CNS. The finding that Hb9;delta;Eh retains significant
activity in rescuing lateral position and robo3 expression indicates
that Hb9 may regulate robo2 and robo3 via distinct mechanisms,
perhaps involving different transcriptional cofactors or
intermediate targets. In support of this hypothesis, hb9 overexpression
in the ap neurons can induce robo2, but not robo3.
These data raise the intriguing possibility that Hb9's ability to
regulate robo2 and robo3 via different mechanisms contributed
to the diversification of their expression patterns in the CNS.
Determining how Hb9 and Nkx6 regulate their effectors will be
key to achieving a complete understanding of how these
conserved transcription factors control changes in cell morphology
and axon pathfinding during development. Of note,
Hb9 mutant mice exhibit defects in a subset of motor nerves,
including the phrenic and intercostal nerves, which are also
affected in Robo mutants. It will be of great interest
to determine if despite the vast divergence in the evolution of
nervous system development between invertebrates and vertebrates,
Hb9 or Nkx6 has retained a role for regulating Robo receptors
across species (Santiago, 2014).
Frazzled (Fra) and deleted in colorectal cancer (Dcc) are homologous receptors that promote axon attraction in response to netrin. In Drosophila, Fra also acts independently of netrin by releasing an intracellular domain (ICD) that activates gene transcription. How neurons coordinate these pathways to make accurate guidance decisions is unclear. This study shows that the ADAM metalloprotease Tace cleaves Fra, and this instructs the switch between the two pathways. Genetic manipulations that either increase or decrease Tace levels disrupt midline crossing of commissural axons. These conflicting phenotypes reflect Tace's function as a bi-directional regulator of axon guidance, a function conserved in its vertebrate homolog ADAM17: while Tace induces the formation of the Fra ICD to activate transcription, excessive Tace cleavage of Fra and Dcc suppresses the response to netrin. It is proposed that Tace and ADAM17 are key regulators of midline axon guidance by establishing the balance between netrin-dependent and netrin-independent signaling (Zang, 2022).
This study reports a conserved role for Tace and ADAM17 in coordinating different receptor signaling outputs. In the context of commissural axon guidance, strong biochemical and genetic evidence is provided demonstrating that Tace functions in both the canonical netrin-dependent Fra pathway and the non-canonical netrin-independent Fra pathway. Specifically, this study has shown that in the non-canonical pathway, Tace cleaves Fra to induce the formation of a transcriptionally active ICD fragment that regulates the expression of Fra's target gene comm. In the canonical pathway, overexpression of Tace results in excessive cleavage of Fra that inhibits the receptor's ability to respond to netrin. Because both Fra pathways are essential for proper midline attraction, such bi-directional regulation by Tace could assist the neurons in orchestrating appropriate signaling outputs. Thus, a model is proposed in which the levels and/or activity of Tace must be correctly regulated to ensure that Fra signals effectively. When the levels or activities of Tace are low, the commissural neuron favors the netrin-dependent canonical pathway to promote midline crossing. In contrast, when Tace is activated, the commissural neuron decreases its responsiveness to netrin and switches to the non-canonical Fra pathway to regulate gene transcription. Together, these two pathways are maintained in a tightly controlled balance and cooperate to facilitate commissural axon midline crossing. Importantly, this study also demonstrated a clear functional similarity between Tace and ADAM17, arguing that ADAM17-dependent cleavage of Dcc likely allows for a similar segregation of receptor signaling outputs in vertebrate systems (Zang, 2022).
Previous studies have revealed that ADAM-dependent proteolysis can impinge on downstream signaling pathways of guidance receptors in many ways. First, ADAM cleavage can terminate receptor signaling by reducing receptor surface levels. Second, ADAM cleavage can facilitate association between the receptor and its downstream effector proteins.Third, ADAM cleavage can physically separate receptor-ligand complexes to switch adhesion to repulsion. Previous studies have demonstrated that both of Fra's vertebrate homologs, Dcc and neogenin, are also ADAM substrates, yet the physiological significance and the precise mechanisms of how proteolysis affects downstream signaling of these receptors remain unclear. This study has shown that in Drosophila, Tace induces the formation of both Fra ECDs and ICDs and is required for the expression of Fra's transcriptional target gene comm. Unlike the mechanisms described above, the current data establish a distinct pathway in which an ADAM can activate an axon guidance receptor by initiating subsequent γ-secretase cleavage to induce transcriptional activity of the receptor.
It remains to be seen whether the same non-canonical mechanism is conserved for ADAM17 and Fra's vertebrate homologs Dcc and neogenin. In Drosophila commissural neurons, γ-secretase cleaves Fra to generate an ICD fragment, allowing it to translocate to the nucleus to regulate the transcription of comm (Zang, 2022).
Previous studies have demonstrated that the neogenin ICD is present in the nucleus in both chick retinal ganglion cells and zebrafish embryos. In addition, in vitro evidence from cell lines suggests that both neogenin and Dcc ICDs can localize to the nucleus to modulate gene transcription, suggesting that similar non-canonical pathways exist for Dcc and neogenin as well. This study has shown that conditional removal of Adam17 disrupts midline crossing in vivo. If the only function of ADAM17 at the vertebrate midline is to downregulate netrin and Dcc signaling, it would be expected to see increased crossing when ADAM17 levels are reduced. Thus, in vertebrates, ADAM17 behaves similarly to Tace in Drosophila to positively regulate midline crossing. This observation supports a potential role for ADAM17 in activating the transcriptional activities of Dcc at the vertebrate midline (Zang, 2022).
While Dcc is arguably the most likely substrate, it should be noted that analysis in embryonic mouse spinal cords does not exclude the possibility that ADAM17 functions by regulating other substrates. One possibility is neogenin, which, in addition to netrin, also binds to its canonical ligand repulsive guidance molecule (RGM) with much higher affinity. Cleavage of neogenin by ADAM17 desensitizes axons to RGM, which abolishes its repulsive effects on neurite outgrowth. Importantly, neogenin contributes to commissure formation in the mouse spinal cord by binding and facilitating Dcc and netrin signaling through its ECDs (Zang, 2022).
Thus, it is unlikely that neogenin is the primary ADAM17 substrate, as shedding of its ECDs should have deleterious effects on midline guidance, which is contrary to what wsd observed in Adam17 cKO embryos. Another possible substrate is the repulsive receptor Robo1. While it has not been determined whether Robo1 is a substrate of ADAM17, it is possible that cleavage of Robo1 inhibits its repulsive function, which could explain the reduced commissure formation observed in Adam17 cKO embryos. One would predict that, as a result, overexpression of Tace or ADAM17 should inhibit Robo1-mediated repulsion, which is contrary to what was observed in Drosophila. This again argues against Robo1 as the primary substrate mediating the phenotypes observed in Adam17 cKO embryos (Zang, 2022).
The data suggest that Tace and ADAM17 are strictly regulated to achieve a precise balance of Fra and Dcc signaling outputs. What are the mechanisms involved in commissural neurons to modulate Tace and ADAM17 expression and/or activity? In principle, this regulation could occur either at the metalloprotease level or at the substrate level. First, the surface expression or the activity of Tace and ADAM17 could be regulated. This study has demonstrated that tace and Adam17 transcripts are highly expressed in the embryonic CNS of both invertebrates and vertebrates. In Drosophila, the expression of Tace mRNA and protein does not appear to be temporally controlled, suggesting that Tace activity is likely to be regulated post-translationally. Indeed, a number of molecules have been identified as regulators of ADAM17 activity, including tissue inhibitor of metalloproteinases-3 (TIMP-3) which suppresses ADAM17 catalytic activity, and the adapter proteins iRhom1 and iRhom2, which are involved in ADAM17 maturation and stability.
Thus, it would be interesting to explore the potential roles of TIMP-3 and the iRhoms and their Drosophila orthologs during axon guidance (Zang, 2022).
Alternatively, regulation of Tace and ADAM17 function could occur at the substrate level. Ligand binding could induce conformational changes in the substrate to facilitate its association with the metalloprotease, or an interacting protein could bind to the substrate to block association with the metalloprotease. Previous work has shown that netrin does not activate the transcriptional activity of Fra,sugge sting that an alternative ligand may exist for the non-canonical pathway. In mouse cortical neurons, leucine-rich repeats and immunoglobulin-like domains 2 (Lrig2) bind to neogenin to inhibit premature ADAM17 cleavage (Zang, 2022).
It remains to be seen whether similar mechanisms regulate Fra and Dcc cleavage.
Tace and ADAM17 coordinate the canonical and non-canonical pathways
ADAMs can facilitate a switch in responses to guidance cues. For example, ADAM10 and ADAM17 cleave Neuropilin-1 to regulate proprioceptive axon responsiveness to Sema3A.
In addition, ADAM10 cleaves ephrinA5 to convert EphA3-ephrinA5-mediated adhesion to repulsion.
It is proposed that Tace and ADAM17 can instruct neurons to coordinate the canonical and non-canonical pathways. It is important to point out that, instead of producing opposite signaling outcomes, the canonical and non-canonical pathways both promote midline crossing of commissural axons, which suggests that the two pathways cooperate instead of competing. This distinguishes the current model from existing mechanisms, but at the same time inevitably poses the question: how does the neuron decide between the two pathways? It is speculated that both pathways could be engaged in the same cell but are separated spatially and/or temporally. The canonical pathway is activated at the tip of the axon, where Fra responds to netrin to locally regulate the cytoskeleton (Zang, 2022).
It is possible that the non-canonical Fra pathway is activated in the soma instead, where the cleaved Fra ICDs are within close proximity to the nucleus. This is supported by the observation that both endogenous Tace and overexpressed Tace are almost exclusively detected in the cell soma but not on the axons. In addition, the two pathways could be activated by distinct ligands, so that they are controlled at different developmental time points. Given the diverse roles of ADAM family metalloproteases in development and disease, continued investigation into their regulation and mechanism of action will undoubtedly offer important biological insights (Zang, 2022).
While this study demonstrated that Tace/ADAM17 is a key regulator of Fra/Dcc signaling and controls midline axon guidance, this study has its limitations. First, tace zygotic mutants in Drosophila show midline crossing defects with low penetrance, which is likely the result of a maternal effect. Yet it was not possible to definitively test this by generating maternal zygotic tace mutants, due to developmental arrest potentially linked to its function in the Notch pathway. Second, it remains to be seen if the same non-canonical signaling mechanism is conserved for Dcc in vertebrate systems. Future transcriptomic studies comparing gene expression levels in controls and Dcc cKO embryos or cell lines, or cell lines overexpressing Dcc ICD, could help resolve its role in transcriptional regulation. Third, it is unclear if Tace/ADAM17 is required cell autonomously in commissural neurons or cell non-autonomously in neighboring cells to cleave Fra/Dcc. Generating commissural neuron-specific cKO animals in the future could provide definitive answers. Fourth, the commissure formation deficits observed in Adam17 mutants are not causatively linked to a lack of Dcc cleavage. Finally, it remains to be determined what the biological stimulus that activates the non-canonical pathway is (Zang, 2022).
NADPH oxidases (Nox; see Drosophila Nox) are membrane-bound multi-subunit protein complexes producing reactive oxygen species (ROS) that regulate many cellular processes. Emerging evidence suggests that Nox-derived ROS also control neuronal development and axonal outgrowth. However, whether Nox act downstream of receptors for axonal growth and guidance cues is presently unknown. To answer this question, retinal ganglion cells (RGCs) derived from zebrafish embryos were cultured and these neurons were exposed to netrin-1, slit2 (see Drosophila Slit), and brain-derived neurotrophic factor (BDNF). To test the role of Nox in cue-mediated growth and guidance, Nox was either pharmacologically inhibited or neurons from mutant fish were investigated that are deficient in Nox2. Slit2-mediated growth cone collapse, and axonal retraction was eliminated by Nox inhibition. Though no effect of either BDNF or netrin-1 was seen on growth rates, growth in the presence of netrin-1 (see Drosophila Netrins) was reduced by Nox inhibition. Furthermore, attractive and repulsive growth cone turning in response to gradients of BDNF, netrin-1, and slit2, respectively, were eliminated when Nox was inhibited in vitro. ROS biosensor imaging showed that slit2 treatment increased growth cone hydrogen peroxide levels via mechanisms involving Nox2 activation. The possible relationship between Nox2 and slit2/Robo2 signaling was investigated in vivo. astray/nox2 double heterozygote larvae exhibited decreased area of tectal innervation as compared to individual heterozygotes, suggesting both Nox2 and Robo2 are required for establishment of retinotectal connections. These results provide evidence that Nox2 acts downstream of slit2/robo2 by mediating growth and guidance of developing zebrafish RGC neurons (Terzi, 2020).
Drosophila Robo3 is a member of the evolutionarily conserved Roundabout (Robo) receptor family and one of three Drosophila Robo paralogs. During embryonic ventral nerve cord development, Robo3 does not participate in canonical Slit-dependent midline repulsion, but instead regulates the formation of longitudinal axon pathways at specific positions along the medial-lateral axis. Longitudinal axon guidance by Robo3 is hypothesized to be Slit dependent, but this has not been directly tested. In this study a series of Robo3 variants was created in which the N-terminal Ig1 domain is deleted or modified, in order to characterize the functional importance of Ig1 and Slit binding for Robo3's axon guidance activity. Robo3 is shown to require its Ig1 domain for interaction with Slit and for proper axonal localization in embryonic neurons, but deleting Ig1 from Robo3 only partially disrupts longitudinal pathway formation. Robo3 variants with modified Ig1 domains that cannot bind Slit retain proper localization and fully rescue longitudinal axon guidance. These results indicate that Robo3 guides longitudinal axons independently of Slit, and that sequences both within and outside of Ig1 contribute to this Slit-independent activity (Carranza, 2023).
Early endosomes are essential for regulating cell signalling and controlling the amount of cell surface molecules during neuronal morphogenesis. Early endosomes undergo retrograde transport (clustering) before their homotypic fusion. Small GTPase Rab5 is known to promote early endosomal fusion, but the mechanism linking the transport/clustering with Rab5 activity is unclear. This study showed that Drosophila Strip is a key regulator for neuronal morphogenesis. Strip knockdown disturbs the early endosome clustering, and Rab5-positive early endosomes become smaller and scattered. Strip genetically and biochemically interacts with both Glued (the regulator of dynein-dependent transport) and Sprint (the guanine nucleotide exchange factor for Rab5), suggesting that Strip is a molecular linker between retrograde transport and Rab5 activation. Overexpression of an active form of Rab5 in strip-mutant neurons suppresses the axon elongation defects. Thus, Strip acts as a molecular platform for the early endosome organization that has important roles in neuronal morphogenesis (Sakuma, 2014).
The formation of neuronal connections requires the precise guidance of developing axons toward their targets. In the Drosophila visual system, photoreceptor neurons (R cells) project from the eye into the brain. These cells are grouped into some 750 clusters comprised of eight photoreceptors or R cells each. R cells fall into three classes: R1 to R6, R7, and R8. Posterior R8 cells are the first to project axons into the brain.Using a microarray-based molecular screen as a starting point, this study identified the early and transient expression of Robo3 in R8 growth cones. Loss of Robo3 demonstrated a specific axon guidance choice point at an early stage of optic lobe innervation. In the absence of Robo3, posterior R8 growth cones inappropriately extend across Slit-expressing glial cells joining axon fascicles of the C+T lobula neurons, instead of remaining alongside the glial process as they extend into the lamina. This early repulsive function of Robo3 plays a crucial role in segregating axons and thereby contributes to the orderly assembly of columnar units comprising the fly visual system (Pappu, 2011).
The microarray data, coupled with antibody staining and the identification of a robo3-Gal4 enhancer trap, identified Robo3 as an R8-specific guidance receptor. Robo3 expression is transient in the R8 growth cone and prolonging Robo3 expression in R8 axons results in defects in R8 targeting. The microarray analysis suggests that restricted Robo3 expression during early stages of R8 differentiation occurs downstream of the transcription factors Sens and Run. However, the expression of both Sens and Run persists beyond the expression of Robo3. For example, during mid- to late-pupal development, Sens regulates both targeting of R8 axons to their final target layer in the medulla and the expression of R8-specific opsins. Therefore, other mechanisms must exist to control the expression of Robo3 in R8 (Pappu, 2011).
The importance of the tightly regulated expression of Robo receptors is emerging as a central theme in axon guidance. Indeed, previous studies have revealed a set of discrete posttranslational mechanisms controlling Robo functions both in vertebrates and invertebrates. For example, commissural axons in the fly embryo express Robo1 transcripts before crossing the midline, but Robo1 protein in these axons is sequestered into endosomes by the action of Commissureless protein, thereby preventing precocious repulsion from the midline and, thus, allowing these axons to cross. Subsequent up-regulation of Robo1 prevents them from recrossing. In vertebrates, alternative splicing of a divergent Robo receptor Rig1/Robo3, perhaps coupled with translational regulation, governs the switch from midline attraction to repulsion (Pappu, 2011).
The regulated expression of Robo3 in the R8 photoreceptors is similar to its expression in the chordotonal neurons in the embryonic peripheral nervous system. Sens is activated downstream of Ato in both chordotonal and in R8 neurons, suggesting that a conserved transcriptional program regulates Robo3 expression in these neurons. In a broader sense, these findings raise the possibility that conserved regulatory cassettes exist, which link specific transcriptional hierarchies controlling neuronal differentiation with specific constellations of downstream guidance receptors controlling wiring specificity (Pappu, 2011).
Posterior-most R8 neurons face at least three different guidance choices as they extend from the eye disk into the developing optic lobe. These early choices have a profound effect on later aspects of visual system assembly. First, R8 axons must navigate to the posterior of the eye disk and enter the optic stalk. This process is facilitated, in part, by retinal basal glial cells at the posterior edge of the eye disk. If glial cells are displaced anteriorly, R-cell axon fascicles project away from the optic stalk rather than toward it. Although it seems likely that this directional choice relies upon R8, it is not known whether posterior growth requires R8-specific functions or whether all retinal neurons are endowed with this function (Pappu, 2011).
Second, R8 axons from each ommatidium must possess molecular mechanisms to retain their individuality. As the R8 axons extend down the optic stalk they form a tight fascicle. Fasciculation is transient, however, because R8s defasciculate as they exit the optic stalk. R8 defasciculation relies on two cell surface receptors, Flamingo (Fmi) and Golden Goal (Gogo) that are expressed in the R8 growth cones as they exit the optic stalk. Fmi and Gogo mediate repulsive interactions between R8 axons, and thus play a key role ensuring that columns remain as separate modules. These repulsive interactions between R8 axons of adjacent columns also explain why axons from later-born (anterior) R8 neurons are not affected in robo33 mutant optic lobes; only the posterior R8 axons traverse through the optic lobe with access to the glial cell boundaries that separate them from the C+T lobular neuron axons (Pappu, 2011).
Third, in this article it was demonstrated that posterior R8 axons rely upon Robo3 to prevent inappropriate fasciculation with C+T lobula neurons. This process requires early, transient, and specific expression of Robo3 in R8 growth cones and is likely to require the reciprocal expression of Slit in the glial cells that these posterior R8 axons encounter when they enter the optic lobe. Thus, the posterior R8 axons are unique because they navigate a choice point that is not encountered by later arriving, more anterior R8 axons. The robo33 mutant phenotype described in this study is reminiscent of the loss of another Ig receptor encoded by the irregular chiasm/roughest (irre-C) locus. Whether IrreC acts in the same molecular pathway as Robo3 and, indeed, whether it acts in photoreceptor growth cones or lamina neurons is not known (Pappu, 2011).
In summary, Fmi, Gogo, and Robo3 play crucial roles in R8s in regulating fascicle organization, which provides the structural basis for columnar organization of the visual system. Although Fmi and Gogo mediate interactions between axons of the same class of cells (R8s), Robo3 prevents axons from one class of neurons (R8s) from inappropriately associating with a different class of axons (C+T lobular neurons) projecting into the same neuropil along a different pathway. Given the cellular complexity of columns (e.g., medulla columns comprise more than 50 axons from many different neuronal subclasses) and the stereotyped organization of axons and synaptic connections within them, it is speculated that many additional cell-surface proteins must act in a coordinated fashion in space and time to promote the orderly assembly of columnar units (Pappu, 2011).
Does Robo3 function in the R8 rely on Slit? This is indeed the most parsimonious model for Robo3 function in R8. Slit expression is detected around glial cells separating the posterior R8 growth cones from C+T lobula neurons. Although R8 projection defects seen in slit mutants are similar to those seen in robo3 mutants, they are more severe. In contrast to robo3, slit mutant optic lobes are extremely disorganized, arguing that Slit has a broader role in neuropil organization. Indeed, mutants deficient in all three Robo proteins exhibit cell migration defects, which largely phenocopy the loss of Slit. It has been proposed that Slit provides a repellent function in the optic lobe preventing cell migration between cell populations in the lamina and lobula. However, the residual Robo3 function in the robo31 hypomorphic allele available for study at that time masked the robo3 phenotypes uncovered in this study. Thus, although slit mutants uncover a broader role for Slit-Robo signaling in many aspects of optic lobe development and patterning, the unique robo3 mutants described in this study uncover a specific role for the Robo3 receptor in R8 axon guidance (Pappu, 2011).
The repulsive role of Robo3 in R8 neurons in response to locally secreted Slit is proposed in this study to be analogous to the role of Robo1 in the guidance of ipsilateral longitudinal pioneer axons in the ventral nerve cord. Robo1 is expressed on the growth cones of ipsilateral pioneer axons and prevents these axons from crossing the midline in response to Slit secreted from midline glia. In contrast, Robo3 is not expressed in the growth cone of commissural and longitudinal pioneer axons and is dispensable for the midline crossing during the development of the embryonic ventral nerve cord. Thus, the function of Robo3 in posterior R8s is analogous to the function of Robo1 in embryonic ipsilateral pioneers. However, analyses of knock-in mutants indicate that Robo1 and Robo3 must be sensitive to cell-type-specific regulatory functions. Robo1 has a unique role in midline repulsion of ipsilateral pioneers as it cannot be functionally replaced by Robo2 or Robo3. In contrast, as reported in this study, either Robo1 or Robo2 can functionally replace Robo3 in the R8s. Thus, repulsive signaling downstream of ipsilateral pioneers in the embryo is dependent on unique structural features of the Robo1 protein, but repulsion of posterior R8 axons does not depend on unique structural features of Robo3. Instead it is the unique and context-specific expression of Robo3 that allows it to determine R8 axon guidance and in a broader context function in the orderly assembly of a subset of columnar elements in the visual circuit (Pappu, 2011).
The Vav proteins are guanine exchange factors (GEFs) that trigger the activation of the Rho GTPases in general and the Rac family in particular. While the role of the mammalian vav genes has been extensively studied in the hematopoietic system and the immune response, there is little information regarding the role of vav outside of these systems. This study reports that the single Drosophila vav homolog is ubiquitously expressed during development, although it is enriched along the embryonic ventral midline and in the larval eye discs and brain. The role that vav plays during development was analyzed by generating Drosophila null mutant alleles. The results indicate that vav is required during embryogenesis to prevent longitudinal axons from crossing the midline. Later on, during larval development, vav is required within the axons to regulate photoreceptor axon targeting to the optic lobe. Finally, it was demonstrated that adult vav mutant escapers, which exhibit coordination problems, display axon growth defects in the ellipsoid body, a brain area associated with locomotion control. In addition, this study showed that vav interacts with other GEFs known to act downstream of guidance receptors. Thus, it is proposed that vav acts in coordination with other GEFs to regulate axon growth and guidance during development by linking guidance signals to the cytoskeleton via the modulation of Rac activity (Malartre, 2010).
Vav members are key regulators of the Rho GTPases and the Rac proteins in particular. However, although many studies have implicated the Rac proteins in controlling several aspects of axon growth and guidance during development, understanding of vav function in these processes is far more primitive. This is quite surprising given that all vav members are expressed in neural tissues in mammals. Recently, analysis of postnatal vav2/vav3 -deficient mice has revealed abnormal retinogeniculate projections. This study demonstrates that in Drosophila vav is required for axon growth and guidance at embryonic, larval, and pupal stages. Hence, these data strengthen the role of vav in multiple aspects of axogenesis during development (Malartre, 2010).
During the formation of the embryonic central nervous system of Drosophila, the neurons send out axons that project either ipsilaterally or contralaterally to form the complex axonal lattice. A small number of neurons project ipsilaterally as they receive repulsive signals from the midline glia and never cross the midline, while most neurons project contralaterally, cross the midline, and form the commissures. In vav2/vav3 mutant mouse brains, ipsilateral but not contralateral projections are affected. These results are consistent with the current data showing that Drosophila vav is required to regulate proper ipsilateral axon projection, as in vav mutant embryos the most medial longitudinal axons occasionally cross the midline when they should not. These fascicles are particularly sensitive to perturbations in axon guidance mechanism and cross the midline whenever repulsive signaling is altered, suggesting that Vav might participate in the regulation of repulsive signaling from the midline (Malartre, 2010).
This study also demonstrated that vav is required during subsequent larval development in regulating photoreceptor axon targeting to the optic lobe. This is again consistent with the finding that vav2/vav3 mutant mice display abnormal projections of axons connecting the retinal cells to the brain, suggesting that the role of vav in mediating axon guidance decisions is conserved between species. Interestingly, vav function in photoreceptors (R cells) seems to be more important than in the embryonic CNS. Indeed, this study found that in 100% of the larvae, R cell axons projected aberrantly to the lamina and the medulla target regions, while only 14% of vav mutant embryos displayed guidance defects. Finally, it was shown that later, during metamorphosis, vav is required once more for the correct formation of the ellipsoid body, one of the central brain structures. The ellipsoid body, in a majority of vav mutant adult brains, does not close properly and remains ventrally opened, most likely reflecting defects in the growth of the axons forming the ellipsoid body rather than guidance errors. Interestingly, the ellipsoid body has been involved in the control of locomotion, and vav mutant adults display strong locomotion defects. Opened ellipsoid bodies have also been found in ciboulot mutants. However these mutants do not display locomotion defects, suggesting that a disruption of the ellipsoid body alone is not sufficient to produce the locomotion phenotype observed in vav mutants. This implies that vav might also be required to regulate other aspects of axogenesis, in addition to the ones identified in this study (Malartre, 2010).
In summary, these results show that vav is required reiteratively throughout life to regulate different axogenesis events, including axon growth and guidance (Malartre, 2010).
During larval development, R cell axon targeting to the optic lobe is controlled, on the one hand, by some genes that are acting within the axons themselves, and on the other hand, by some genes that are sending signals to the axons from the glia to guide them. MARCM experiments clearly demonstrate a role for vav within the R cell axons to regulate their projections. This is also the case in mammals, where Vav2 is highly expressed in the growth cones of cultured neurons where it is required to control guidance (Malartre, 2010).
Interestingly, in Drosophila, the GEF Trio has been shown to activate Rac, which in turn activates Pak, which is recruited to the membrane by Dock. These proteins participate in a signal transduction pathway that plays an essential role during photoreceptor axon guidance. Vav also acts via Rac in photoreceptors, and vav and trio interact genetically. Thus, in this context, it is tempting to speculate that like Trio, Vav could also contribute to the precise spatial control of Pak activity. In this scenario, the combination of signals via Vav, Trio, and Dock would allow growth cones to integrate multiple guidance signals (Malartre, 2010).
Vav function in the axons could be to regulate the intracellular trafficking of guidance receptors through the activation of Rac. In mammals, for instance, vav2 has been proposed to be required in axons downstream of ephrin signaling for proper axon guidance. In this case, when ephrins bind their Eph receptors, Vav becomes transiently activated upon phosphorylation and promotes local Rac-dependent endocytosis of the ephrin/Eph complex, a key event in axonal repulsion. In Drosophila however, mutations in Eph surprisingly show no obvious axon guidance defects in the photoreceptor axons targeting to the optic lobe nor in the embryonic CNS. This suggests that in Drosophila, vav would need to be acting downstream of other guidance signals besides Eph (Malartre, 2010).
In conclusion, it is proposed that Vav, after being activated by signaling receptors, could be required to stimulate Rac proteins to participate in the regulation of axon growth and guidance during development (Malartre, 2010).
The Drosophila genome contains 22 GEFs. At least nine of them are expressed in the CNS, five of which are thought to be Rac activators. Why are there several Rac GEFs acting in the nervous system? A possible explanation is that the different GEFs might be activated in response to distinct guidance cues, thus triggering Rac-dependent specific cellular responses. For instance, beside its function in longitudinal axon growth, Trio has been involved in promoting commissure formation through its interaction with the attractive Netrin receptor Frazzled. Furthermore, another GEF, Sos, has been proposed to mediate Rac activation downstream of the Robo receptor to control axon repulsion at the midline. In this scenario, Vav, Sos, and Trio could coexist and be activated in response to different guidance molecules to control distinct aspects of axon guidance during the formation of the CNS (Malartre, 2010).
In another scenario, different set of GEFs could also act redundantly to activate Rac proteins to a certain level, or at precise time points or in specific subcellular locations, allowing a unique cellular response. In fact, this study has shown that the loss of both vav and sos function enhances dramatically the individual midline guidance phenotypes, suggesting that vav and sos can act redundantly in a common pathway. Similarly, the phenotype of the vav;trio double mutant in the nervous system, both at the midline and along the longitudinal axons, is more severe than the single mutants. In addition, while mutations in either vav or trio do not show any obvious defects outside the nervous system despite their widespread expression, elimination of both results in gross morphological defects. This indicates that both genes can act redundantly in vivo in different tissues and suggests that vav and trio are the main regulators of Rac activity (Malartre, 2010).
A final explanation for the existence of different rac GEFs is that they could preferentially activate a particular Rac. There are three highly related rac genes in Drosophila, rac1, rac2, and mtl, and it has been suggested that Rac1 and Rac2 are preferred substrates of Trio. By performing a similar epistasis analysis, this study has shown that in photoreceptor cells Vav activates preferentially Rac1 and Rac2. The fact that vav and trio show similar substrate specificities could explain why these two genes were found to be redundant during embryogenesis (Malartre, 2010).
In conclusion, although the vav family has been mainly implicated in the hematopoietic system and immune response, new roles are beginning to emerge for these genes. The fact that vav is required for axon growth and guidance at different stages of development suggests that it could be playing a multiplicity of functions in response to diverse signals. The existence of various protein-protein interaction domains in Vav represents a means of integrating Rac activities. These results also suggest that vav function must overlap with that of other Rac modulators. Having isolated mutations in the Drosophila vav gene will help elucidate not only the role of this GEF during neural development but also the molecular mechanisms underlying general remodeling of the embryonic and adult nervous systems (Malartre, 2010).
Kinesin-1 can slide microtubules against each other, providing the mechanical force required for initial neurite extension in Drosophila neurons. This sliding is only observed in young neurons actively forming neurites and is dramatically downregulated in older neurons. The downregulation is not caused by the global shutdown of kinesin-1, as the ability of kinesin-1 to transport membrane organelles is not diminished in mature neurons, suggesting that microtubule sliding is regulated by a dedicated mechanism. This study has identified the "mitotic" kinesin-6 Pavarotti (Pav-KLP) as an inhibitor of kinesin-1-driven microtubule sliding. Depletion of Pav-KLP in neurons strongly stimulated the sliding of long microtubules and neurite outgrowth, while its ectopic overexpression in the cytoplasm blocked both of these processes. Furthermore, postmitotic depletion of Pav-KLP in Drosophila neurons in vivo reduced embryonic and larval viability, with only a few animals surviving to the third instar larval stage. A detailed examination of motor neurons in the surviving larvae revealed the overextension of axons and mistargeting of neuromuscular junctions, resulting in uncoordinated locomotion. Taken together, these results identify a new role for Pav-KLP as a negative regulator of kinesin-1-driven neurite formation. These data suggest an important parallel between long microtubule-microtubule sliding in anaphase B and sliding of interphase microtubules during neurite formation (Del Castillo, 2014).
Previous work showed that microtubule sliding by kinesin-1 drives initial neurite outgrowth in Drosophila neurons and that sliding is downregulated as neurons mature. This paper, has demonstrated that the 'mitotic' kinesin Pav-KLP functions as a negative regulator of interphase microtubule sliding both in Drosophila S2 cells and in Drosophila neurons. Knockdown of Pav-KLP stimulated microtubule sliding, producing longer axons, while overexpression of Pav-KLP inhibited both sliding and neurite outgrowth. Increased length of axons after Pav-KLP depletion was also observed in vivo in Drosophila. Therefore, Pav-KLP attenuates neurite outgrowth by downregulation of kinesin-1-powered microtubule-microtubule sliding (Del Castillo, 2014).
Pav-KLP and its orthologs (members of the kinesin-6 family) were originally identified as essential components for central spindle assembly and cleavage furrow formation. Pav-KLP depletion induces defects in morphology of the mitotic spindle at telophase and failure to recruit contractile ring components. However, it has been demonstrated that CHO1/MKLP1, the mammalian ortholog of Pav-KLP, has an additional function in neurodevelopment. CHO1/MKLP1 plays a role in establishing dendrite identity in differentiated neurons. Depletion of CHO1/MKLP1 induces progressive loss of dendrites. It has concluded that CHO1/MKLP1 organizes microtubules in dendrites by transporting short minus-end-distal microtubule fragments into the dendrites. More recent work has revisited the role of CHO1/MKLP1 in developing neurons and suggested that CHO1/MKLP1 can regulate neurite outgrowth. Depletion of CHO1/MKLP1 increased transport of short microtubule fragments. The current data are in agreement with the idea that Pav-KLP regulates formation of neurites. However, the mechanisms reported in this study are clearly different from the results obtained by the mammalian studies in two significant aspects. First, this study has shown that the reorganization of microtubules required for neurite formation is driven by kinesin-1. Second, the current visualization technique clearly demonstrates that microtubules in developing Drosophila neurons are moved as long polymers. It is possible that the differences between the results and the mammalian study can be explained by different models (Drosophila versus mammalian neurons). A more attractive idea is that similar mechanisms work in both systems, but further work is required to understand the role of kinesin-1 in neurite outgrowth in mammalian neurons (Del Castillo, 2014).
Interestingly, work by several groups has shown that proteins that function together with kinesin-6 in the cytokinesis pathway could also regulate neuronal morphogenesis. For example, Tumbleweed or Ect2/Pebble/RhoGEF depletion increases the extent of neurite outgrowth, suggesting that Tumbleweed and RhoGEF control neurite outgrowth through actin reorganization. However, the current results demonstrate that the primary regulator of neurite outgrowth is kinesin-6 family member Pav-KLP, the essential partner of Pebble and Tumbleweed. Furthermore, the effect of Pav-KLP on process formation is independent of actin or small GTPases (although more subtle effects of Tumbleweed or Ect2 on the actin cytoskeleton in developing neurons cannot be completely excluded). Indeed, a recent work concluded that an actin-signaling pathway regulated by the Centralspindlin complex controls protrusive activity required for directional neuronal migration (Del Castillo, 2014).
The original idea that mitotic motors regulate cytoplasmic microtubules in neurons suggested that microtubule arrays in neurons are established by mechanisms that are analogous to those that organize the mitotic spindle. Supporting this idea, it was demonstrated that inhibition of other mitotic motors, e.g., kinesin-5, affected the axon length Advancing this concept, this paper proposes that Pav-KLP/kinesin-6 directly regulates cytoplasmic microtubule arrangement by crosslinking them. It has been shown that loss-of-function mutations on ZEN-4/MKLP1, the C. elegans form of Pav-KLP, produce longer spindles, suggesting that kinesin-6 motors inhibit sliding of microtubules against each other during anaphase B. If this is indeed the case, the current results suggest an important functional similarity between the molecular mechanisms of cell division and process formation in neurons. While anaphase B is driven in part by microtubule-microtubule sliding powered by bipolar kinesin-5 and negatively regulated by kinesin-6 (mammalian MKLP1/C. elegans Zen-4/Drosophila Pav-KLP), the initial formation of neurites requires microtubule-microtubule sliding by kinesin-1 and, similar to anaphase B, is negatively regulated by kinesin-6. Thus, kinesin-6 motors together with other components of the Centralspindlin complex can function as general brakes of microtubule-microtubule sliding during both cell division and postmitotic neurite formation (Del Castillo, 2014).
The fact that microtubule sliding is inhibited by Pav-KLP in mature, but not young, neurons suggests that Pav-KLP itself is temporally regulated. One potential mechanism that could affect the ability of Pav-KLP (MKLP-1) to regulate microtubule sliding is Pav-KLP phosphorylation. Phosphorylation of Ser710 in MKLP-1 (Ser743 in Drosophila Pav-KLP) has been shown to promote its binding to protein 14-3-3, preventing MKLP-1 from clustering on microtubules. Future studies using phosphomimetic variants of Pav-KLP may help to test this mechanism (Del Castillo, 2014).
In many eukaryotic cells, directed molecular transport occurs along microtubules. Within neuronal axons, transport over vast distances particularly relies on uniformly oriented microtubules, whose plus-ends point towards the distal axon tip (anterogradely polymerizing, or plus-end-out). However, axonal microtubules initially have mixed orientations, and how they orient during development is not yet fully understood. Using live imaging of primary Drosophila melanogaster neurons, this study found that, in the distal part of the axon, catastrophe rates of plus-end-out microtubules were significantly reduced compared to those of minus-end-out microtubules. Physical modelling revealed that plus-end-out microtubules should therefore exhibit persistent long-term growth, while growth of minus-end-out microtubules should be limited, leading to a bias in overall axonal microtubule orientation. Using chemical and physical perturbations of microtubule growth and genetic perturbations of the anti-catastrophe factor p150, which was enriched in the distal axon tip, it was confirmed that the enhanced growth of plus-end-out microtubules is critical for achieving uniform microtubule orientation. Computer simulations of axon development integrating the enhanced plus-end-out microtubule growth identified in this studya with previously suggested mechanisms, that is, dynein-based microtubule sliding and augmin-mediated templating, correctly predicted the long-term evolution of axonal microtubule orientation as found in the experiments. This study thus leads to a holistic explanation of how axonal microtubules orient uniformly, a prerequisite for efficient long-range transport essential for neuronal functioning (Jakobs, 2022).
Mechanisms governing a neuron's regenerative ability are important but not well understood. This study has identified Rtca (RNA 3'-terminal phosphate cyclase) as an inhibitor of axon regeneration. Removal of Rtca cell-autonomously enhanced axon regrowth in the Drosophila CNS, whereas its overexpression reduced axon regeneration in the periphery. Rtca along with the RNA ligase Rtcb and its catalyst Archease operate in the RNA repair and splicing pathway important for stress-induced mRNA splicing, including that of Xbp1, a cellular stress sensor. Drosophila Rtca and Archease had opposing effects on Xbp1 splicing, and deficiency of Archease or Xbp1 impeded axon regeneration in Drosophila. Moreover, overexpressing mammalian Rtca in cultured rodent neurons reduced axonal complexity in vitro, whereas reducing its function promoted retinal ganglion cell axon regeneration after optic nerve crush in mice. This study thus links axon regeneration to cellular stress and RNA metabolism, revealing new potential therapeutic targets for treating nervous system trauma (Song 2015).
Failure of damaged axons to regenerate is the primary cause of permanent disabilities after CNS injury and the irreversible neurologic dysfunction of neurodegenerative diseases. The ability of a neuron to regenerate its axon after trauma is governed by the interaction between its intrinsic growth capacity and the local environment. Notwithstanding the discoveries of extracellular factors and intrinsic pathways that reduce the regenerative capacity of axons, effective therapies have not yet emerged because removing the known inhibitory cues only partially restores regeneration, thus indicating the presence of additional inhibitory machineries that remain to be discovered (Song 2015).
Studies using model organisms such as Caenorhabditis elegans have begun to identify new genes important for axon regeneration, illustrating the power of the genetic approach. To identify more factors that control axon regeneration, a Drosophila sensory neuron injury model that exhibits class-specific axon regeneration was established and it was demonstrated that the class IV dendritic arborization (da) neuron is capable of regenerating its axon in the periphery but exhibits limited regrowth inside the CNS, resembling its mammalian counterpart at the phenotypic and molecular levels (Song, 2012). Using this model, a candidate-based genetic screen was performed focusing on axotomy-regulated genes from several organisms and Drosophila Rtca (CG4061), a cellular RNA-processing enzyme with unknown biological function, was identified as an inhibitor of CNS axon regeneration. Furthermore, it was found that Drosophila Archease, a RNA ligase cofactor, functions downstream of Rtca as a pro-regeneration factor. Rtca and Archease are components of the RNA repair and splicing pathway, and they regulate the unconventional mRNA splicing of Xbp1, a stress sensor. Thus, Xbp1 acts as a substrate, readout and downstream effector for the regulation of axon regeneration by the RNA repair and splicing pathway (Song 2015).
To assess axon regeneration, a previously described protocol (Song, 2012). Briefly, with a two-photon laser, the axons of class IV da neurons (labeled with pickpocket (ppk)-CD4tdGFP) was severed in the ventral nerve cord (VNC) of second-instar larvae 48 h after egg laying (AEL), the degeneration of the remaining axons was confirmed after 1 d (72 h AEL) and their regeneration was assessed after 2 more days (120 h AEL). Using this model, the effect was measured of RtcaNP5057, an insertional loss of function (LOF) allele with a P-element inserted in the 5'-UTR, disrupting mRNA splicing and reducing transcript expression. Compared to wild types, which showed limited regrowth, new axons regrew extensively from the retracted axon stems and extended into the commissure region, forming elaborate branches and reconnected commissure segments in RtcaNP5057 larvae. Similar phenotypes were seen in transheterozygotes of RtcaNP5057 over a deficiency line, Df(1)BSC718, that lacks the Rtca locus and in a Rtca deletion allele, RtcaΔ, generated from imprecise excision of RtcaNP5057. Even stronger phenotypes were seen in RtcaΔmat, in which both the zygotic and maternal transcripts were removed. RtcaNP5057 is homozygous viable and fertile, so these larvae were derived from homozygous mutant mothers. The mothers of RtcaNP5057/Df(1)BSC718 transheterozygotes and RtcaΔ mutants were heterozygous for the wild-type allele and may provide maternal wild-type Rtca transcripts. The fact that RtcaΔmat mutants, in which both the zygotic and maternal transcripts were removed, showed a stronger phenotype than RtcaΔ zygotic mutants confirmed the maternal effect. Thus, the phenotype of RtcaNP5057 mutants compared to RtcaNP5057/Df(1)BSC718 transheterozygotes and RtcaΔ mutants is likely stronger because no wild-type maternal transcripts were provided to RtcaNP5057 mutants. The function of Drosophila Rtca is cell autonomous, as its RNA interference knockdown in class IV da neurons (ppk-Gal4>RtcaRNAi) but not in glial cells (repo-Gal4>RtcaRNAi) recapitulated the enhancement of regeneration. The regeneration phenotype was further quantified by assessing the following metrics, as described previously (Song, 2012): regeneration percentage, terminal branching and commissure regrowth. The enhancement of regeneration is unlikely to be due to developmental defects of axon outgrowth because, first, the overall axon patterning of class IV da neurons in the uncut VNC is grossly normal and second, reducing Rtca function in RtcaΔ mutants or transheterozygotes of RtcaNP5057 over Df(1)BSC718 or via Rtca RNAi in class IV da neurons did not result in obvious defects of axon terminal patterning in the VNC (Song 2015).
Tests were performed to see whether reducing Drosophila Rtca function would trigger a regenerative response in neurons normally incapable of regeneration by severing their axons in Rtca mutants. Indeed, Rtca removal in class III da neurons (labeled with 19-12-Gal4>CD4tdGFP, repo-Gal80), which unlike class IV da neurons did not regrow their axons that were severed in the periphery, elicited substantial regeneration in RtcaΔmat mutants and after RNAi knockdown of Rtca specifically in class III da neurons, leading to significant increases in the regeneration percentage, regeneration index and regeneration length (Song 2015).
Conversely, overexpression of Rtca in class IV da neurons (ppk-Gal4>Rtca) mildly reduced their regenerative potential in the peripheral nervous system (PNS). In wild-type class IV da neurons, which regenerated about 74% of the time, new axons extended beyond the lesion site and followed the axonal track. In contract, Rtca overexpression caused the incidence of regeneration to be reduced to 48% and the length of the new axons to be significantly shortened as well. These data indicate that Drosophila Rtca is an inhibitor of axon regeneration: not only does its removal cell-autonomously enhance axon regeneration in the CNS and enable regeneratively incompetent neurons such as class III da neurons to regrow their axons in the PNS, its overexpression in regeneratively competent neurons impedes axon regeneration in the periphery (Song 2015).
The inhibitory function of Drosophila Rtca is, furthermore, not limited to sensory neurons. Rtca overexpression in motor neurons also suppressed motor axon regeneration after nerve crush, as demonstrated by the reduced elaboration of growth cones (Song 2015).
The expression pattern of Drosophila Rtca was examined via two approaches. First, the P-element inserted in Rtca 5'-UTR (RtcaNP5057) contains Gal4 in the same orientation as Rtca and thus can allow inference of Rtca expression via a UAS reporter. It was found that the Rtca-Gal4>CD4tdGFP reporter colocalized with the class IV da neuron marker ppk-CD4tdTomato, confirming its presence in class IV da neurons. Although Rtca-Gal4 expression was observed in other tissues in the PNS and VNC, the analyses indicate that Drosophila Rtca functions cell autonomously in neurons to inhibit axon regeneration (Song 2015).
A polyclonal antibody was generated against Drosophila Rtca. The protein was present in wild-type but not in RtcaΔmat null class IV and class III da neurons, and was enriched in the nucleus. Drosophila Rtca was also present in other types of multidendritic neurons (Song 2015).
To begin to understand the mechanisms underlying Drosophila Rtca's role in regeneration, attempts were made to determine how it genetically interacts with the known axon regeneration regulators Pten (phosphatase and tensin homolog) and the cytoskeletal regulator Rac1 GTPase. Deletion of Pten, a negative regulator of the mammalian target of rapamycin (mTOR) pathway, has been shown to increase CNS axon regeneration in both mammals and flies. Overexpression of Rtca in a Pten hypomorphic mutant background (PtenMGH6; ppk-Gal4>Rtca) or overexpression of Pten in Rtca null mutants (RtcaΔ; ppk-Gal4>Pten) largely abolished the enhancement of axon regeneration as seen in the VNC in PtenMGH6 or RtcaΔ mutants. This suggests that Rtca and Pten are likely to function in parallel pathways. Notably, double mutation of Rtca and Pten (RtcaNP5057; PtenMGH6) did not further improve regeneration, as compared to Rtca mutation alone, indicating the presence of additional brakes on regeneration. The regeneration phenotype in Rtca mutants appeared to be comparable to if not stronger than that in PtenMGH6 mutants or that seen with Akt overexpression (ppk-Gal4>Akt) (Song 2015).
Because Rac is required for regenerative axon outgrowth in C. elegans, this study overexpressed Rac1 in class IV da neurons (ppk-Gal4>Rac1). An increase was found in the number of axons initiating the regenerative response in the VNC but not in terminal branching or commissure regrowth; that is, there was a partial improvement in regeneration. Conversely, overexpressing a dominant negative (DN) form of Rac1 abolished the enhancement of CNS axon regeneration seen in Rtca null mutants (RtcaΔmat; ppk-Gal4>Rac1DN), whereas Rac1DN overexpression alone in class IV da neurons did not result in obvious axon regeneration defects in the PNS. It thus seems likely that Rac1 functions downstream of Rtca in a pathway that converges on regulation of the cytoskeleton (Song 2015).
Rtca is a RNA processing enzyme that possesses RNA-3'-phosphate cyclase activity and catalyzes the ATP-dependent conversion of a 3' phosphate to a 2',3'-cyclic phosphodiester at the end of RNA molecules (Genschik, 1997). The RNA 2',3'-cyclic phosphate ends are important in RNA metabolism—for example, as intermediates during RNA repair by ligases (Popow, 2011; Remus, 2013). Rtcb (RNA 2',3'-cyclic phosphate and 5'-OH ligase) represents a new type of RNA ligase that joins 2',3'-cyclic phosphate and 5'-OH RNA ends to yield a 3'-5' phosphodiester splice junction. Specifically, Rtcb is known to possess cyclic phosphodiesterase activity, which hydrolyzes the 2',3'-cyclic phosphate to a 3'-phosphate, as well as ligase activity, which then joins the RNA 3'-phosphate to a 5'-OH RNA end. In addition, the specificity and efficacy of Rtcb's ligase activity can be enhanced by Archease (Desai, 2014; Popow, 2014), which is a small acidic protein conserved among Eukarya, Bacteria and Archaea. In Escherichia coli, RtcA and RtcB are encoded in a single operon, suggesting that they might cooperate to provide a healing and sealing function in an RNA repair pathway (Tanaka, 2011). In one scenario, healing would refer to the restoration of ligatable 2',3'-cyclic phosphate ends in the event of the inciting RNA damage directly generating RNA 3'-phosphates, or of the 2',3'-cyclic phosphate products of RNA transesterification being further processed to a 3'-phosphate by a 2',3'-cyclicphosphodiesterase. However, this model cannot readily be reconciled with the subsequent finding that RtcB readily joins 3'-phosphate to 5'-OH ends or 2',3'-cyclic phosphate to 5'-OH ends (Song 2015).
Therefore, the exact relationship between RtcA and RtcB remains undetermined. Notably, the RtcBA operon in E. coli is regulated by the σ54 coactivator RtcR, suggesting that the RNA repair functions are induced in response to cellular stress. Although the biological function of Rtca remains unknown, the enzyme is speculated to act in some aspect of cellular RNA processing . Taking into account these findings and the observation that loss of Rtca function enhances axon regeneration, it is hypothesized that the Rtca-Archease-dependent RNA repair and splicing pathway regulates axon regeneration. Specifically, it is speculated that axon injury triggers a type of cellular stress leading to RNA damage and splicing, producing RNA 3'-phosphates that need to be processed and rejoined by the Rtcb ligase, which is catalyzed by Archease. Because Rtca converts RNA 3'-phosphate to 2',3'-cyclic phosphate, it can slow the ligation process and impede regeneration. Consequently, silencing Rtca promotes axon regeneration. Following this reasoning, the role of Archease in axon regeneration was investigated (Song 2015).
To determine the role of the Drosophila Archease in axon regeneration, regeneration of class IV da neuron axons in the periphery was examined. To maximize the phenotype, the PNS axon injury protocol was modified as described previously (Song, 2012): axotomy was induced at 72 h AEL, degeneration was confirmed at 96 h AEL and regeneration was assayed at 120 h AEL. The Archease (CG6353) LOF mutant allele ArcheasePBc01013, which is an insertional allele with a P-element inserted into the 5'-UTR disrupting its mRNA splicing and eliminating Archease transcripts. Unlike in wild-type neurons, which exhibited substantial regrowth of their severed axons, axon regeneration was significantly impaired in ArcheasePBc01013 neurons, as revealed by a significant drop of the regeneration percentage, regeneration index and regeneration length. This phenotype was confirmed in transheterozygotes of ArcheasePBc01013 over either of the two deficiency lines, Df(3R)ED6076 or Df(3R)BSC678, that lack the Archease locus. The ArcheasePBc01013 mutation is larval lethal. ArcheasePBc01013 mutants and ArcheasePBc01013/DfED6076 and ArcheasePBc01013/DfBSC678 transheterozygotes showed similar phenotypes, suggesting that ArcheasePBc01013 is likely an amorphic allele. The function of Archease is required cell-autonomously, as class IV da neuron-specific knockdown of Archease (ppk-Gal4>ArcheaseRNAi) but not glial cell knockdown (repo-Gal4>ArcheaseRNAi) was sufficient to phenocopy the regeneration failure. Moreover, loss of function of both Rtca and Archease (RtcaNP5057; ArcheasePBc01013) completely abolished the axon regeneration-promoting effect in the VNC seen in RtcaNP5057 mutants, producing many retracted or stalled axon stems. This epistasis analysis indicates that Archease is a pro-regeneration factor downstream of Rtca and that they act in opposing ways to regulate axon regeneration (Song 2015).
What might be the RNA substrates processed by this Drosophila Rtca-Archease-dependent RNA repair and splicing pathway for the regulation of axon regeneration? This study investigated X-box binding protein 1 (Xbp1) as a candidate substrate for three reasons. First, cellular stress such as endoplasmic reticulum (ER) stress triggers an adaptive intracellular signaling cascade known as the unfolded protein response (UPR). One main branch of the UPR is the activation of Ire1, which cleaves Xbp1 pre-mRNA in the cytoplasm, converting the unspliced Xbp1μ, a putative transcriptional repressor, into the unconventionally spliced Xbp1s by eliminating an intron (26 nucleotides in mammals, 23 in flies) that changes the open reading frame of the third exon, resulting in a new protein that acts as a transcriptional activator (Yoshida, 2001). Xbp1s directly activates ER stress target genes to facilitate refolding and also degradation of misfolded proteins (Ron, 2007). Second, the RNA ligase Rtcb and its cofactor Archease are involved in the unconventional splicing induced by the UPR, and Archease is required for the splicing of the Xbp1 mRNA (Jurkin, 2014). Third, loss of xbp1 function in C. elegans results in severely reduced axon regeneration (Nix, 2014). To determine the function of Xbp1 in axon regeneration in the PNS and CNS, a mutant allele, Xbp1k13803, was used that has a P-element inserted into its 5'-UTR, thus reducing transcripts (Ryoo, 2007). Axon regeneration in the periphery was mildly reduced in these mutants. This defect was stronger in transheterozygotes of Xbp1k13803 over a deficiency line, Df(2R)BSC484, that lacks the Xbp1 locus, suggesting that Xbp1k13803 is likely a hypomorphic allele. Class IV da neuron-specific (ppk-Gal4>Xbp1RNAi) but not glia-specific (repo-Gal4>Xbp1RNAi) RNAi of Xbp1 reproduced the impairment of regeneration, indicating it functions cell-autonomously. Moreover, double mutation of Rtca and Xbp1 (RtcaNP5057; Xbp1k13803) dampened the enhancement of CNS axon regeneration seen in RtcaNP5057 mutants, indicating that Xbp1 is indeed a pro-regeneration factor downstream of Drosophila Rtca. Consistent with these lines of evidence, overexpression of the spliced form Xbp1s in class IV da neurons significantly enhanced axon regeneration in the VNC, and it also promoted axon regeneration in the periphery when overexpressed in class III da neurons. The observations that Rtca; Xbp1 double mutation did not completely eliminate the enhanced regeneration phenotype in Rtca mutants and that Xbp1s overexpression led to milder enhancement of regeneration as compared to Rtca LOF, suggest that additional substrates contribute to regeneration regulation (Song 2015).
To directly assess the nonconventional splicing of Xbp1 mRNA in vivo, a heat-shock model was used. Fly larvae of various genotypes underwent a 40°C heat shock and the abundance of Xbp1 splice variants was assessed using semiquantitative RT-PCR. The Xbp1s/Xbp1μ ratio was then quantified. Heat-shock induced the expression of the spliced form, Xbp1s. In contrast to the enhanced expression of Xbp1s in Rtca mutants, Xbp1s levels were greatly reduced in Archease LOF mutants. Double mutants of Rtca and Archease resembled Archease mutants in the reduction of Xbp1 splicing. Taken together, these data indicate that Drosophila Rtca and Archease in the RNA repair and splicing pathway negatively and positively regulate the stress-induced Xbp1 mRNA splicing, respectively, so that Xbp1 acts as a readout and effector for the regulation of axon regeneration (Song 2015).
Having established the role of Rtca in axon regeneration in Drosophila, the study went on to determine whether its function is evolutionarily conserved in mammals. The expression pattern of the mammalian ortholog of Drosophila Rtca was examined in vitro. Antibodies raised against human RTCA recognized rat Rtca in the cell bodies and processes of cultured hippocampal neurons. Moreover, the expression of Rtca transcripts in the dorsal root ganglion (DRG) in vivo increased progressively throughout development, reaching the highest level in adults. Using quantitative RT-PCR, it was found that Rtca transcript levels in the DRG were significantly reduced following lesion of the sciatic nerve peripherally, but not lesion of the central axon branch of DRG neurons with a spinal cord hemisection. Since the peripheral processes of DRG neurons are capable of regeneration, whereas their central axons that project into the spinal cord fail to regrow after injury, the selective suppression of Rtca following peripheral injury supports the hypothesis that the persisting expression of Rtca is inhibitory to axon regeneration in the CNS. Furthermore, in agreement with the overexpression phenotype in flies, overexpression of Rtca in cultured hippocampal neurons reduced axon complexity and markedly reduced proximal axonal branching without affecting total axon length, indicating an inhibitory function of Rtca (Song 2015).
It was next asked whether knocking out Rtca during development enhances axon regeneration of adult mouse retinal ganglion cells (RGCs) in vivo. For this purpose, a mutant allele was generated with a lacZ cassette inserted after the third exon to disrupt splicing and reduce transcription (to ~19%), thereby generating RtcalacZ_loxP (RtcaIns/Ins) mutant mice. Rtca protein level was also reduced to ~18% in the mutants, suggesting this is a hypomorphic allele. Homozygous RtcaIns/Ins mice were born, although at less than the Mendelian ratio. By adulthood, there were no obvious differences in RGC number or RGC axon morphology among mutant (RtcaIns/Ins), heterozygous (RtcaIns/+) and wild-type (Rtca+/+) animals. Since the lacZ cassette is inserted into the Rtca locus, it can be used as a reporter for examining Rtca expression. β-Galactosidase staining was observed in the RGC layer of RtcaIns/+ mice but not in Rtca+/+ littermates . Moreover, β-galactosidase immunostaining in RtcaIns/+ mice showed distinct expression of the lacZ reporter in NeuN+ neurons in the retina, which was absent in Rtca+/+ littermates, indicating that Rtca is indeed expressed in RGCs. To assess RGC axon regeneration, optic nerve crush was perfored in RtcaIns/Ins, RtcaIns/+ and Rtca+/+ littermate mice at two developmental time points, postnatal day (P) 35 and 2-3 months old, and the extent of axon regeneration was measured in the optic nerve after 2 weeks or 3 weeks, respectively. RtcaIns/+ and Rtca+/+ mice injured at P35 did not exhibit substantial axon regrowth beyond the crush site, whereas RtcaIns/Ins mutant mice showed a substantial increase in the number of regenerating axons at various distances from the injury site, with some regenerating axons extending over 1.5 mm beyond the crush site. Mice operated on at 2-3 months old showed a milder regeneration enhancement phenotype. In these animals, curving, turning and looping of axons were observed, indicative of new axon growth, and the furthest distance that axons traveled beyond the injury site was about 3.5 times longer in the mutants. The crush site was further marked by the presence of ED1 staining, which labels infiltrating macrophages. Whereas axons rarely penetrated beyond the ED1+ region in sibling controls, a large number of axons were seen hundreds of microns beyond the ED1+ region in RtcaIns/Ins mutants. Reducing Rtca function did not affect RGC survival after injury, confirming that this increase in regenerating axons was not secondary to an increase in RGC numbers. The finding that reducing Rtca expression increased the regenerative potential of adult RGCs thus provides evidence for a potentially conserved role of Rtca as an anti-regeneration factor (Song 2015).
These findings reveal an important role of the RNA repair and splicing pathway in regulating the intrinsic axon regeneration potential in response to PNS and CNS injury in Drosophila. Rtca and Archease integrate the injury signals triggered by axotomy and lead to the activation of downstream effectors such as the stress response cascade involving Xbp1 splicing, affecting the ability of a neuron to regenerate. Axon injury has been suggested as a cellular stress, and the mTOR pathway, a potential determinant of neuronal regeneration competence, could be inactivated under stress conditions such as hypoxia or DNA damage (Lu, 2014). Notably, Xbp1 splicing has been observed in RGCs after optic nerve injury and forced activation of Xbp1 promotes RGC survival (Hu, 2014). This work implicates proper splicing of Xbp1 as also important for axon regeneration in Drosophila (Song 2015).
Moreover, recent work in C. elegans also suggests the involvement of stress response pathways, such as heat-shock, hypoxia and UPR, in axon regeneration (Nix, 2014). However, how the injury signal is relayed to the stress response is unclear. This work identifies a missing link and implicates the Rtca-Archease-dependent RNA metabolism machinery as a regeneration regulator. A priori, axonal injury could either signal directly to the stress pathways, which then recruit Rtca-Archease, or alternatively, Rtca and Archease may represent injury response elements upstream of the stress pathways. The results showed that the Xbp1-dependent UPR pathway acts downstream of Rtca-Archease in controlling axon regeneration, and the remaining question is whether and how it impinges on other stress pathways, such as hypoxia or DNA damage. It will be important in future studies to identify other substrates, in addition to Xbp1, that are modified by Rtca-Archease, and to search for response genes downstream of Xbp1. This study raises the prospect of manipulating Rtca, Archease and Xbp1 as potential therapeutic interventions for treating nervous system injury (Song 2015).
As a first step to determining whether the Rtca pathway may have an evolutionarily conserved function in axon regeneration, this study has examined CNS axon regeneration after optic nerve crush in a hypomorphic mouse mutant allele of Rtca and evidence was obtained suggesting that this is indeed the case. The enhancement of RGC axon regeneration phenotype in the Rtca mutant is modest as compared to that seen in Pten, Klf4 or Socs3 knockouts. This may be due to the residual Rtca function in this hypomorphic allele or to developmental compensation. Future experiments using mammalian injury models to examine the Rtca null allele and to assess other components of the RNA repair and splicing pathway are therefore warranted to further define its potential role in axon regeneration (Song 2015).
Axonal growth and targeting are fundamental to the organization of the nervous system, and require active engagement of the cytoskeleton. Polymerization and stabilization of axonal microtubules is central to axonal growth and maturation of neuronal connectivity. Studies have suggested that members of the Tubulin Polymerization Promoting Protein (P25alpha/TPPP) family are involved in cellular process extension. However, no in vivo knockout data exists regarding its role in axonal growth during development. This study reports the characterization of Ringmaker (Ringer; CG45057), the only Drosophila homolog of long p25alpha proteins. Immunohistochemical analyses indicate that Ringer expression is dynamically regulated in the embryonic CNS. ringer null mutants show cell misplacement, and errors in axonal extension and targeting. Ultrastructural examination of ringer mutants revealed defective microtubule morphology and organization. Primary neuronal cultures of ringer mutants exhibit defective axonal extension, and Ringer expression in cells induced microtubule stabilization and bundling into rings. In vitro assays showed that Ringer directly affects tubulin, and promotes microtubule bundling and polymerization. Together these studies uncover an essential function of Ringer in axonal extension and targeting through proper microtubule organization (Mino, 2016).
Precise axon growth and guidance rely on microtubule polymerization, stabilization and bundling. These processes are central in establishing neuronal connectivity. Various proteins affecting microtubule dynamics have been characterized in the context of process extension. Proteins containing p25α domains are expressed in embryonic and postnatal brains, and are known to alter microtubule dynamics. However, the majority of studies do not address their relevance during early development. Using in vivo and in vitro studies, this study addressed the previously uncharacterized function of Drosophila TPPP (Ringer), the only long p25α-containing protein in Drosophila, and its importance in neuronal development (Mino, 2016).
Through mRNA and protein localization, this work uncovers that Ringer is present in the nervous system and that its expression is variable and tightly modulated in the embryonic CNS midline. Evidence is provided that Ringer is necessary for correct nervous system development. Loss of Ringer results in soma misplacement, and defects in axonal extension and guidance in agreement with neuron-specific knockdown experiments showing similar defects. That loss of Ringer results in axonal disruption is strengthened by the findings of knockdown studies in vitro and in zebrafish, which have shown TPPPs have an effect on process extension. Similarly, ringer has been identified as a neuronal outgrowth modifier candidate (Mino, 2016).
ringer mutants exhibit phenotypic variability. Initially, it was supposed that these differences were due to a contribution of maternal Ringer, a suspicion arising from experiments involving deficiency lines. However, all ringer-mutant embryos analyzed were from homozygous stocks, which rules out this possibility. Phenotypic variance could also arise owing to compensation by other proteins. For instance, TPPP has been suggested to bundle microtubules in manner similar to that of Tau. In Drosophila neurons, Tau knockdown only shows exacerbated neuronal degeneration when combined with futsch mutations. It is hypothesized that Ringer acts in a manner similar to Tau. Additionally, ringer-null mutants exhibit decreased organism viability. Lack of Ringer, as in the case of Tau, leads to reduced viability but not complete lethality (Mino, 2016).
These studies determined that Ringer, like mammalian TPPPs, is able to regulate microtubule dynamics. This is evidenced in vivo by microtubule disruption at segmental nerves in ringer mutants and supported by primary culture studies in which changes in Ringer translate into changes in acetylated tubulin. Ringer is likely to have a conserved stabilizing and bundling function similar to that of mammalian TPPPs. Cell culture experiments too suggest this, as they show Ringer can protect microtubules from depolymerization in addition to altering microtubule architecture, further underscoring a stabilizing function. Furthermore, purified Ringer data show that no other external factors are necessary to induce changes in microtubule dynamics. Thus, this work demonstrates that Ringer alone is sufficient to induce higher rates of microtubule polymerization as well as bundling and stabilization (Mino, 2016).
This work provides evidence that Ringer regulates microtubule changes necessary for axonal development. Ringer is expressed along the axon in primary neurons, and at cellular margins and membrane-ruffle areas in S2 cells, a location concomitant with process growth. Moreover, axon extension and growth cone advancement rely on microtubules. Consequently, in ringer mutants exhibiting axonal stalling and breaks, phenotypes might be representative of lower microtubule polymerization rates that result from lack of Ringer function. This is supported by evidence that Ringer is necessary endogenously for proper axonal extension, and by micrographs showing axonal microtubule disruption in ringer mutants. Surprisingly, both ringer-mutant and -overexpressing neurons exhibit delayed axonal extension. Although defects observed in Ringer overexpression in vivo could be explained by the contribution of Ringer from surrounding cells, overexpression in primary neurons and in vivo Eve-positive neurons also results in soma placement and axonal phenotypes, revealing that there is a cell autonomous Ringer function (Mino, 2016).
The similar phenotypes produced by Ringer loss and gain of function appear counterintuitive. However, in vitro data show that Ringer has the ability to promote microtubule polymerization and bundling. Studies have revealed that modest microtubule overstabilization leads to an overall decrease in dynamics. It is possible that Ringer overexpression stabilizes microtubules sufficiently to prevent axonal advancement, whereas in ringer mutants, axons delay advancement owing to lower tubulin polymerization. Additionally, Ringer loss might lead to depolymerization due to higher susceptibility to severing agents. Perhaps there are Ringer concentration thresholds, post-translational modifications or other factors that decide in favor of a specific function (Mino, 2016).
Conversely, the CNS axon mistargeting observed in ringer mutants might be an indirect result of delays in axonal extension. During embryogenesis, VNC midline neurons extend their axons as they migrate ventrally. In mutants, axons from misplaced neurons might not extend at a normal rate, causing them to miss cues resulting in guidance defects. Additionally, axon guidance defects are repeatedly accompanied by severe neuronal misplacement, suggesting these two phenotypes are linked to migration errors. Alternatively, guidance phenotypes might result from a function of Ringer in growth cone directional movement through differential microtubule stabilization. Thus, it is postulated that during axonal development, Ringer regulates microtubule stabilization that is necessary for correct spatial distribution and polymerization to direct growth (Mino, 2016).
Interestingly, none of the phenotypic rescue experiments yielded a full recovery. Besides FASII embryonic phenotypic rescue, other attempts proved modest at best. These differences are not due to changes in transgene expression but from diverging protein level requirements between systems. Moreover, FASII rescue measurements were performed relative to the integrity of the combined neuronal connections, whereas single-cell measurements were made unhindered by environmental cues. Another possibility is that Ringer is necessary in surrounding cells, such as lateral glia, and that the antibodies are not robust enough to detect Ringer expression in such cells. If this is the case, elav-GAL4-mediated rescue, which expresses Ringer in all neurons and lateral glia at early stages, would be the only driver able to rescue phenotypes. Although these observations do not discard the notion of a function for Ringer in other cells that could influence development, they support the idea of an endogenous cell autonomous Ringer function in neurons (Mino, 2016).
In summary, this work has demonstrated that Ringer contributes to development in the regulation of axonal extension. Ringer was shown to be sufficient to promote microtubule stabilization, bundling and polymerization and that its absence is likely to affect axonal microtubule dynamics, leading to extension delays, mistargeting and, consequently, abnormal neural development (Mino, 2016).
Axonal branching is one of the key processes within the enormous complexity of the nervous system to enable a single neuron to send information to multiple targets. However, the molecular mechanisms that control branch formation are poorly understood. In particular, previous studies have rarely addressed the mechanisms underlying axonal bifurcation, in which axons form new branches via splitting of the growth cone. This study demonstrates that DISCO Interacting Protein 2 (DIP2) is required for precise axonal bifurcation in Drosophila mushroom body (MB) neurons by suppressing ectopic bifurcation and regulating the guidance of sister axons. DIP2 localizes to the plasma membrane. Domain function analysis revealed that the AMP-synthetase domains of DIP2 are essential for its function, which may involve exerting a catalytic activity that modifies fatty acids. Genetic analysis and subsequent biochemical analysis suggested that DIP2 is involved in the fatty acid metabolization of acyl-CoA. Taken together, these results reveal a function of DIP2 in the developing nervous system and provide a potential functional relationship between fatty acid metabolism and axon morphogenesis (Nitta, 2016).
Developmental axon pruning is essential for wiring the mature nervous
system, but its regulation remains poorly understood. This study shows
that the endosomal-lysosomal pathway regulates developmental pruning of
Drosophila mushroom body γ neurons. The UV radiation
resistance-associated gene (Uvrag) functions together with all core
components of the phosphatidylinositol 3-kinase class III (PI3K-cIII; see Phosphotidylinositol 3 kinase 59F)
complex to promote pruning via the endocytic pathway. By studying
several PI3P binding proteins, this study found that Hrs, a subunit of the ESCRT-0 complex, required for multivesicular body (MVB)
maturation, is essential for normal pruning progression. Thus, the
existence of an inhibitory signal that needs to be downregulated is
hypothesized. Finally, the data suggest that the Hedgehog receptor,
Patched, is the source of this inhibitory signal likely functioning in a
Smo-independent manner. Taken together, this in vivo study demonstrates
that the PI3K-cIII complex is essential for downregulating Patched via
the endosomal-lysosomal pathway to execute axon pruning
(Issman-Zecharya, 2014).
Neuronal remodeling is an essential step of nervous system
development in both vertebrates and invertebrates. One mechanism used to
remodel neuronal circuits is by the elimination of long stretches of
axons in a process known as axon pruning. With a few exceptions, the
current dogma is that axon pruning of long stretches of axons occurs via
local axon degeneration while axon pruning of short stretches occurs via
retraction. While in some cases remodeling is directly affected by
experience or neural activity, in cases of stereotypical pruning the
identity of the axon that is destined to be pruned does not depend on
experience or neural activity. Because of mechanistic similarities to
Wallerian degeneration and dying back neurodegenerative diseases,
understanding the molecular mechanisms of axon pruning should result in
a broader insight into axon fragmentation and elimination during
development and in disease (Issman-Zecharya, 2014).
The neuronal remodeling of the Drosophila mushroom body (MB) during
development is a unique model system to study the molecular aspects of
axon pruning. The stereotypic temporal and spatial occurrence of MB axon
pruning combined with mosaic analyses provide a platform to perform
genetic screens and molecular dissections of these processes in
unprecedented resolution. The MB is comprised of three types of neurons
that are sequentially born from four identical neuroblasts per
hemisphere. Out of the three MB neuronal types, only the γ neurons
undergo axon pruning, indicating that the process is cell-type specific.
During the larval stage, γ neurons project a bifurcated axon to
the dorsal and medial lobes. At the onset of metamorphosis, the
dendrites of the γ neurons as well as specific parts of the axons
are eliminated by localized fragmentation in a process that peaks at
about 18 hr after puparium formation. Subsequently, γ neurons
undergo developmental axon regrowth, which is distinct from initial axon
outgrowth, to occupy the adult specific lobe (Issman-Zecharya, 2014).
Axon pruning of MB γ neurons depends on the cell-autonomous
expression of the nuclear steroid hormone receptor, ecdysone receptor B1
(EcR-B1). The expression of EcR-B1 is regulated by at least three
distinct pathways: the cohesin complex, the TGF-β pathway, and a
network of nuclear receptors comprised of ftz-f1 and Hr39.
While expression of EcR-B1 is required for pruning, it is not sufficient
to drive ectopic pruning either in γ neurons or in other MB
neurons that do not undergo remodeling. This raises two possible
nonmutually exclusive scenarios: (1) additional molecules are required
to initiate pruning and (2) an inhibitory signal needs to be attenuated
in the MB for pruning to occur. Additionally, the ubiquitin pathway is
also cell-autonomously required in γ neurons for pruning, but the
target that must be ubiquitinated remains unknown. Thus, while
understanding of the cellular sequence of events culminating in the
elimination of specific axonal branches is quite detailed, understanding
of the molecular mechanisms remains incomplete (Issman-Zecharya, 2014).
In a forward genetic screen, this study identified a cell-autonomous
role for the UV radiation resistance-associated gene (UVRAG) in MB
γ neuron pruning. UVRAG was originally identified based on its
ability to confer UV resistance to nucleotide excision repair deficient
cells. It was later shown to function as a tumor suppressor gene deleted
in various types of cancers including colon and gastric carcinomas. UVRAG interacts with Atg6 (also known as
Beclin1), another tumor suppressor gene, and together they promote
autophagy in vitro. Their tumor suppression capabilities
were first attributed to their autophagy-promoting function. However, a
mutant form of UVRAG isolated from colon carcinomas promoted autophagy
normally in cell culture. Both UVRAG and Atg6 are subunits in the
phosphatidylinositol 3-kinase class III (PI3K-cIII) complex, involved in
autophagy and endocytosis. Recent studies have found that UVRAG mediates
endocytosis in an Atg6-dependent manner suggesting that as part of the
PI3K-cIII complex, both proteins regulate various aspects of vesicle
trafficking. Two studies have recently
identified new and seemingly unrelated functions for UVRAG in regulating
DNA repair in response to UV-induced damage and ER to Golgi
trafficking. Finally, an in vivo study has shown that UVRAG
affects organ rotation in Drosophila by regulating Notch endocytosis in
what seemed to be an Atg6-independent manner. A unifying
understanding of the various aspects of UVRAG physiological function in
vivo is still lacking. Likewise, although the PI3K-cIII complex has been
extensively studied and implicated in autophagy, cytokinesis and
endocytosis, its physiological
roles during the normal course of development are not known
(Issman-Zecharya, 2014).
This study reports that UVRAG and the PI3K-cIII complex mediate the
endosome-lysosome degradation of Ptc to promote axon pruning.
Furthermore, the results suggest that Ptc represses pruning via a Smo-
and Hh-independent manner. This study provides evidence for the
existence of a pruning inhibitory pathway originating at the membrane of
MB neurons (Issman-Zecharya, 2014).
This study shows that the endosomal-lysosomal pathway is
cell-autonomously required for developmental axon pruning of mushroom
body (MB) γ neurons. Genetic loss-of-function experiments indicate
that UVRAG, a tumor suppressor gene previously linked to both
endocytosis and autophagy, promotes pruning as part of the
phosphatidylinositol 3-kinase class III (PI3K-cIII) complex and that
UVRAG is required in MB neurons for the formation of
phosphatidylinositol 3-phosphate (PI3P). The
ESCRT-0 complex, which is recruited to the PI3 moiety on
endosomal membranes, is required for pruning, indicating that endosome
to multivesicular body maturation is critical for the normal progression
of axon pruning and suggesting that it involves receptor downregulation.
Genetic loss-of-function and gain-of-function experiments suggest that
downregulation of the Hedgehog receptor Patched (Ptc) by the endocytic
machinery is instrumental in promoting pruning. Finally, the results
suggest that Ptc inhibits pruning in a smo-independent and likely
also hh-independent manner (Issman-Zecharya, 2014).
A recent study suggested that UVRAG is required for Notch endocytosis
during organ rotation in Drosophila in an Atg6-independent manner. While the current study shows that Atg6 is required for pruning,
these seemingly contradicting results can be easily explained by
specific allele differences. The Atg600096 allele, used in
the previous study, is a P element insertion about 100 bp upstream of
the Atg6 gene that does not necessarily create a null allele. Indeed,
this study could also not see any effect of this allele on axon pruning.
This study used an Atg61 null allele created by homologous
recombination resulting in a strong effect on pruning. Furthermore, the
data clearly show that the entire PI3K-cIII complex is required for axon
pruning (Issman-Zecharya, 2014).
The PI3K-cIII complex has been implicated in a wide variety of
membrane trafficking processes ranging from autophagy to endocytosis to
cytokinesis. How the PI3K-cIII is regulated to
participate in these different processes and its physiological roles in
vivo are not well understood. While its role in promoting autophagy is
supported by several studies, deleting
the catalytic unit, Vps34, in sensory neurons does not affect autophagy, but rather endocytosis. Whether this is a common feature of
PI3K-cIII function in neurons remains to be further elucidated. One
attractive hypothesis is that the PI3K-cIII function is determined by
its complex composition. Indeed, it appears that in vitro, UVRAG and
Atg14 are mutually exclusive subunits defining two distinct populations
of the PI3K-cIII complex (Funderburk, 2010; Itakura, 2009). The current
study is consistent with these findings, suggesting that UVRAG may
define an endocytosis-specific PI3K-cIII complex at least in neurons.
The full spectrum of the various PI3K-cIII complexes physiological roles
in vivo remains to be further studied (Issman-Zecharya, 2014).
The PI3K-cIII complex phosphorylates PI to form PI3P on
endosomal membranes. Indeed, this study found that UVRAG is essential
for efficient PI3P formation and that PI3P is
abundant throughout development. It is thus hypothesized that a
PI3P binding protein mediates the effect of UVRAG and the
PI3K-cIII complex on axon pruning. This study has identified Hrs, a
subunit of the ESCRT-0 complex and a PI3P binding protein, as
required for axon pruning. The role of ESCRT-0 in MVB maturation led to
a hypothesis that the endolysosomal pathway is required to downregulate
a signal that originates at the plasma membrane. While signaling can
still occur in the early endosome, it is terminated at the MVB
(Issman-Zecharya, 2014).
What is the identity of this transmembrane protein? Using genetic
loss-of-function and gain-of-function experiments, it is suggested that
Patched (Ptc) is at least one of the transmembrane proteins that is
responsible for mediating the PI3K-cIII pruning defect. Strikingly,
mutating ptc on the background of a Atg6 mutant
significantly suppressed its pruning defect. Furthermore, overexpression
of Ptc in WT brains resulted in a weak to mild pruning defect, depending
on the Gal4 driver. Finally, overexpressing Ptc on the background of an
endosomal defect significantly exacerbated the pruning defect. Together,
these data suggest that Ptc mediates an inhibitory signal that needs to
be attenuated for the normal progression of pruning. Interestingly, Ptc
inactivation by endocytosis followed by lysosomal degradation was
proposed before as a mechanism to activate the Hh pathway. What is the
nature of this signal? Ptc is known to be the Hedgehog (Hh) receptor.
Binding of Hh to Ptc relieves the Ptc-induced suppression of another
transmembrane protein, Smoothened (Smo). Once derepressed, Smo initiates
the intracellular Hh signal that culminates in the expression of
specific nuclear transcription factors. Therefore this study tested the
role of Smo and Hh in developmental axon pruning and, to surprisingly,
demonstrated that both molecules seem to be irrelevant for pruning.
Overexpressing Ptc mutant transgenes within MB neurons to identify the
domains that are important for pruning inhibitions confirmed that Smo
inhibition was not required to inhibit pruning. In contrast, the results
suggest that the ligand binding domain is important. Because the results
suggest that Hh is not required for pruning inhibition, it will be
interesting to investigate in the future what other ligands might bind
to Ptc. In this regard it is interesting to mention that a recent study
has shown that Ptc is a lipoprotein receptor. The precise mechanism of
Ptc action in MB neurons remains to be further elucidated in future
studies (Issman-Zecharya, 2014).
This study has uncovered a role for the endocytic machinery in
downregulating an inhibitory signal that is dependent on Ptc during MB
axon pruning. A recently published study has shown that
the Rab5/ESCRT endocytic pathways are required to downregulate
neuroglian (Nrg) to promote dendrite pruning of sensory neurons in
Drosophila. Both studies highlight that a combination of
both promoting and inhibitory signals during developmental pruning is
likely important to provide fail-safe mechanisms to regulate the process
in a temporal, spatial, and cell-type specific resolution
(Issman-Zecharya, 2014).
The dNab2 polyadenosine RNA binding protein is the D. melanogaster ortholog of the vertebrate ZC3H14 protein, which is lost in a form of inherited intellectual disability (ID). Human ZC3H14 can rescue D. melanogaster dNab2 mutant phenotypes when expressed in all neurons of the developing nervous system, suggesting that dNab2/ZC3H14 performs well-conserved roles in neurons. However, the cellular and molecular requirements for dNab2/ZC3H14 in the developing nervous system have not been defined in any organism. This study shows that dNab2 is autonomously required within neurons to pattern axon projection from Kenyon neurons into the mushroom bodies, which are required for associative olfactory learning and memory in insects. Mushroom body axons lacking dNab2 project aberrantly across the brain midline and also show evidence of defective branching. Coupled with the prior finding that ZC3H14 is highly expressed in rodent hippocampal neurons, this requirement for dNab2 in mushroom body neurons suggests that dNab2/ZC3H14 has a conserved role in supporting axon projection and branching. Consistent with this idea, loss of dNab2 impairs short-term memory in a courtship conditioning assay. Taken together these results reveal a cell-autonomous requirement for the dNab2 RNA binding protein in mushroom body development and provide a window into potential neurodevelopmental functions of the human ZC3H14 protein (Kelly, 2016).
The analysis of dNab2 reveals a number of parallels to another RNA binding protein, dFmr1, which is also an ortholog of a protein lost in heritable intellectual disability, FMRP. As with ZC3H14, FMRP is a ubiquitously expressed protein whose loss leads to defects in brain function. Strikingly, dFmr1 mutant flies show adult MB defects very similar to those described here for dNab2 mutant flies, including thinned/missing α lobes and fused β lobes. Human and Drosophila FMRP/dFmr1 are well-established translational repressors, and while the precise molecular role of ZC3H14 and dNab2 have yet to be determined, the role of these proteins in limiting poly(A) tail length (Pak, 2011; Kelly, 2014) suggests that they could impact the fate of mRNAs in the cytoplasm, perhaps via effects upstream of translation. Finally, the dNab2 ortholog ZC3H14 is highly expressed in hippocampal neurons (Pak, 2011), which are also an important site of FMRP action. These similarities between dNab2/ZC3H14 and dFmr1/FMRP are suggestive of potential links between these RNA binding proteins that warrant further investigation (Kelly, 2016).
Given its proposed molecular role as a Pab, dNab2 is likely to support neurodevelopment and memory via effects on the stability and/or translation of neuronal mRNAs. These roles could be linked such that defects in regulation of RNAs supporting axon projection lead to corresponding defects in memory circuits. Alternatively these phenotypes could reflect a requirement for dNab2 in regulating distinct pools of RNAs involved in each process. The current observations that neuronal RNAi-mediated depletion of dNab2 elicits penetrant effects on locomotor behavior (Pak, 2011) and short-term memory, but comparatively mild effects on α/β-lobe structure (approximately 65% of brains affected), suggests these two phenotypes could stem from effects in different cells and perhaps different target RNAs. Indeed some proteins required for courtship memory act in γ-lobe neurons whose structure is unaffected by dNab2 loss, while other proteins are only required in the α/β-lobes. Future studies will need to define dNab2 target RNAs in groups of brain neurons and assess their roles in axon projection and STM phenotypes that arise upon dNab2 loss (Kelly, 2016).
The RNAs responsible for axonal defects in dNab2 mutant Kenyon cells whose projections the α/β MB lobes are as yet undefined. Although dNab2 is localized to the nucleus at steady-state, the budding yeast Nab2 protein shuttles between the cytoplasm and nucleus, presenting the possibility that dNab2 could impact RNA regulatory processes beyond nuclear processing. Studies have implicated a diverse set of molecules in MB development, including the cell-cell adhesion proteins N-cadherin, Down-syndrome cell adhesion molecule (Dscam), and L1CAM, as well as signaling cascades from Ephrin and Wingless/Wnt signals, providing a number of candidate pathways. Coordinated control of these signals during axon outgrowth, bifurcation, and synapse formation likely requires precise temporal and spatial control of mRNA stability, transport, and translation. The dNab2/ZC3H14 Pab restricts poly(A) tail length in vivo. Thus, the required role in MB axon development could stem from effects on one or more transcript(s) involved in axonal projection and branching. Identifying these target RNAs will require functional assays that define dNab2-regulated transcripts in neurons and physical interaction screens that recover transcripts bound by dNab2. The identity of these transcripts will provide important clues as to how dNab2 influences cellular processes in the fly brain. However, equally important will be determining the fate of these RNAs once bound by dNab2, and testing whether dNab2 primarily influences neuronal gene expression by controlling the nuclear export, stability, transport, or translation of cytoplasmic RNAs, even if its role is primarily restricted to controlling poly(A) tail length in the nucleus. This combined analysis of dNab2 targets and how each is regulated by dNab2 will likely shed considerable light on the role of the dNab2/ZC3H14 protein family in brain development and function (Kelly, 2016).
A central step in organizing the central nervous system development is the growth cone of an axon navigating through guidance cues to reach its specific target. While a great deal of this process has been understood especially in identifying the extracellular guidance cues and their membrane receptors, much less is known about how guidance signals are further relayed to the actin filaments that are central to the mobility of the growth cone. Previous results have shown that Drosophila gene dunc-115 regulates axon projection in the eye and the central nervous system. Furthermore, Dunc-115 has a villin-headpiece (VHD) domain, implying the possibility of binding to actin. To further characterize Dunc-115's functions, this study has identified the isoform Dunc-115L as a possible downstream target in relaying guidance cues further down to the cytoskeleton. Specifically, it was shown that Dunc-115 regulates neural connections in both the eye and the central nervous system in Drosophila and that Dunc-115 contains an actin-binding domain potentially capable of binding to actin filaments. This report shows that Dunc-115 binds to actin via its VHD domain directly, suggesting a possible mechanism for how Dunc-115 relays guidance signals (Roblodowski, 2017).
Injury-induced (Wallerian) axonal degeneration is regulated via the opposing actions of pro-degenerative factors such as SARM1 and a MAPK signal and pro-survival factors, the most important of which is the NAD+ biosynthetic enzyme NMNAT2 that inhibits activation of the SARM1 pathway. This study investigated the mechanism by which MAPK signaling facilitates axonal degeneration. MAPK signaling was shown to promote the turnover of the axonal survival factor NMNAT2 in cultured mammalian neurons as well as the Drosophila ortholog dNMNAT in motoneurons. The increased levels of NMNAT2 are required for the axonal protection caused by loss of MAPK signaling. Regulation of NMNAT2 by MAPK signaling does not require SARM1, and so cannot be downstream of SARM1. Hence, pro-degenerative MAPK signaling functions upstream of SARM1 by limiting the levels of the essential axonal survival factor NMNAT2 to promote injury-dependent SARM1 activation. These findings are consistent with a linear molecular pathway for the axonal degeneration program (Walker, 2017).
A GGGGCC (G(4) C(2)) repeat expansion in the C9orf72 gene is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Although disruptions in axonal transport are implicated in the pathogenesis of multiple neurodegenerative diseases, the underlying mechanisms causing these defects remain unclear. This study performed live imaging of Drosophila motor neurons expressing expanded G(4) C(2) repeats in third-instar larvae and investigated the axonal transport of multiple organelles in vivo. Expression of expanded G(4) C(2) repeats causes an increase in static axonal lysosomes, while it impairs trafficking of late endosomes (LEs) and dense core vesicles (DCVs). Surprisingly, however, axonal transport of mitochondria is unaffected in motor axons expressing expanded G(4) C(2) repeats. Thus,these data indicate that expanded G(4) C(2) repeat expression differentially impacts axonal transport of vesicular organelles and mitochondria in Drosophila models of C9orf72-associated ALS/FTD (Sung, 2022).
Axon regeneration helps maintain lifelong function of neurons in many animals. Depending on the site of injury, new axons can grow either from the axon stump (after distal injury) or from the tip of a dendrite (after proximal injury). However, some neuron types do not have dendrites to be converted to a regenerating axon after proximal injury. For example, many sensory neurons receive information from a specialized sensory cilium rather than a branched dendrite arbor. It was hypothesized that the lack of traditional dendrites would limit the ability of ciliated sensory neurons to respond to proximal axon injury. This hypothesis was tested by performing laser microsurgery on ciliated lch1 neurons in Drosophila larvae and tracking cells over time. These cells survived proximal axon injury as well as distal axon injury, and, like many other neurons, initiated growth from the axon stump after distal injury. After proximal injury, neurites regrew in a surprisingly flexible manner. Most cells initiated outgrowth directly from the cell body, but neurite growth could also emerge from the short axon stump or base of the cilium. New neurites were often branched. Although outgrowth after proximal axotomy was variable, it depended on the core DLK axon injury signaling pathway. Moreover, each cell had at least one new neurite specified as an axon based on microtubule polarity and accumulation of the endoplasmic reticulum. It is concluded that ciliated sensory neurons are not intrinsically limited in their ability to grow a new axon after proximal axon removal (Stone, 2023).
How glia control axon regeneration remains incompletely understood. This study investigated glial regulation of regenerative ability differences of closely related Drosophila larval sensory neuron subtypes. Axotomy elicits Ca(2+) signals in ensheathing glia, which activates regenerative neurons through the gliotransmitter adenosine and mounts axon regenerative programs. However, non-regenerative neurons do not respond to glial stimulation or adenosine. Such neuronal subtype-specific responses result from specific expressions of adenosine receptors in regenerative neurons. Disrupting gliotransmission impedes axon regeneration of regenerative neurons, and ectopic adenosine receptor expression in non-regenerative neurons suffices to activate regenerative programs and induce axon regeneration. Furthermore, stimulating gliotransmission or activating the mammalian ortholog of Drosophila adenosine receptors in retinal ganglion cells (RGCs) promotes axon regrowth after optic nerve crush in adult mice. Altogether, the findings demonstrate that gliotransmission orchestrates neuronal subtype-specific axon regeneration in Drosophila and suggest that targeting gliotransmission or adenosine signaling is a strategy for mammalian central nervous system repair (Wang, 2023).
Axon regeneration failure is a major hurdle for functional recovery after CNS lesion. In addition, Wallerian axon degeneration is associated with neurodegenerative diseases, hence promoting that axon regeneration in these diseases is of great interest. By parallel investigation of closely related Drosophila neuronal subtypes, gliotransmission is proposed as a previously unknown mechanism by which glia could control axon regeneration. These findings expand the existing glial roles in axon regeneration. In a working model that summarizes these Drosophila studies, ensheathing glia actively respond to axotomy and the resulting glial Ca2+ spikes lead to the release of ATP/adenosine. Although adenosine could interact with all sensory neurons, the AdoR expression pattern ensures that C4da, but not C3da, neurons respond to adenosine. This glia-to-neuron signaling then mounts pro-regenerative programs in C4da neurons, including burst firing, Ca2+ spikes, and Ras activity to promote axon regrowth of C4da neurons. Moreover, ectopic AdoR expression or activation of those regenerative programs in C3da neurons is sufficient to induce their axon regeneration (Wang, 2023).
How could axotomy elicit glial Ca2+ signals in Drosophila larvae? Glia are known to express neurotransmitter receptors, allowing them to "listen to" neuronal activity and provide feedback modulation of neuronal activity through gliotransmitters. To assess whether the neuronal activity could have an effect on glial Ca2+ signals, AITC was applied to activate C4da neurons. However, Allyl isothiocyanate (AITC) did not evoke Ca2+ signals in ensheathing glia, arguing against the neuronal activity as a trigger of glial Ca2+ signals. These results support unidirectional signaling from glia to sensory neurons. They also suggest that glial Ca2+ spikes are produced either cell-autonomously or alternatively through a cell-non-autonomous mechanism that involves glial interactions with non-neuronal cells.
Burst firing observed in Drosophila larval C4da neurons is reminiscent of the ectopic discharges of injured dorsal root ganglion (DRG) neurons, the mammalian counterpart of Drosophila larval PNS sensory neurons. As a hallmark of neuropathic pain, the ectopic discharges of DRG neurons are also regulated by gliotransmission. This suggests that burst firing of C4da neurons could reflect nociceptive hypersensitivity after axotomy. Hence, one possible role of gliotransmission could be to sensitize injured C4da neurons. Taken together, gliotransmission appears to be related to both axon regeneration and nociceptive hypersensitivity. Future work is needed to determine whether gliotransmission could regulate DRG axon regeneration (Wang, 2023).
By stimulating Drosophila larval C3da neurons with different firing patterns, this study provides direct evidence that firing patterns, instead of excitability, dictate the axon regenerative strength. A possible mechanism is that neuronal activity patterns are associated with different Ca2+ signals. For example, burst, but not tonic, firing evokes strong Ca2+ signals in C3da neurons, likely due to prolonged depolarization associated with burst firing. Previous studies indicate that the stimulation of neuronal activity promotes mammalian axon regeneration, but the reported effects are variable. One possibility could be that the activity patterns are not controlled in these studies. In the future, it will be of interest to deliver precisely controlled neuronal activity patterns to determine their effects on axon regeneration in other experimental models (Wang, 2023).
It is proposed that the Ca2+ spikes in Drosophila larval C4da neurons are pro-regenerative signal with the following characteristics: they are neuronal-type-specific, correlate with burst firing, exhibit delayed onset, persist throughout axon regeneration, and are evoked by axonal but not dendritic injury. Optogenetic stimulation of C3da neurons starting at ~6 hpa (i.e., after immediate Ca2+ transients) further indicates that Ca2+ spikes are sufficient to induce axon regeneration. Because Ca2+ signals with different spatiotemporal patterns activate diverse neuronal pathways, it is likely that Ca2+ spikes and immediately Ca2+ transients regulate different aspects of axon regeneration. Although immediate Ca2+ transients could regulate growth cone formation, persistent Ca2+ spikes could maintain Ras activity at a high level for sustained axon regrowth. It will be interesting to determine whether axotomy could trigger Ca2+ spikes in other systems, and if so, their roles in axon regeneration (Wang, 2023).
Unlike CNS, mammalian PNS neurons such as DRG can regenerate their axons. Therefore, mammalian CNS and PNS neurons could be broadly defined as two neuronal groups bearing axon regenerative differences, although each group is highly heterogeneous and contains multiple neuronal subtypes. In this regard, the differences between mammalian CNS and PNS are much larger than those between Drosophila C4da and C3da neurons. To explore whether Adora2b signaling could underlie the regenerative differences of mammalian CNS and PNS neurons, the published single-cell RNA-seq dataset was search, but it was found that Adora2b was expressed at low levels in both CNS and DRG neurons. Hence, other mechanisms must exist to explain their regenerative differences. One such mechanism could be related to intrinsic growth abilities. For example, the injury of mouse RGCs causes a reduction of mTOR activity; by contrast, no reduction of mTOR activity was found after the injury of DRG neurons (Wang, 2023).
Glial cells in the mammalian retina could release ATP, glutamate, and D-serine. The mouse study indicates that the stimulation of the gliotransmitter release alone by designer receptor exclusively activated by designer drugs (DREADDs) is ineffective, but combining glial DREADD stimulation and neuronal Adora2b expression greatly enhanced axon regeneration. Because Adora2b is barely expressed in the mouse retina, these results suggest that engineering RGCs to respond to the gliotransmitter adenosine is a prerequisite for the observed effect. Similarly, Drosophila C3da neurons do not express AdoR, and engineering them to respond to adenosine induces axon regeneration. These analyses suggest that stimulation of gliotransmission could promote axon regeneration across species if Adora2b orthologs are expressed in neurons. Notably, Adora2b overexpression in RGCs alone moderately increased axon regeneration, suggesting that extracellular adenosine exists in the injured retina. These results further suggest that in the absence of glial DREADD stimulation, axotomy could trigger basal adenosine release (Wang, 2023).
Adenosine exerts protective functions in different mammalian tissues including the nervous system, by triggering diverse signaling events. Extracellular adenosine is normally low but is rapidly increased after injury. As a classic neuromodulator, adenosine controls essential homeostatic functions such as sleep. However, whether adenosine could play a role in axon regeneration is largely unknown. Pharmacological Adora3 activation is shown to promote neurite outgrowth of cultured RGCs. In the current study, Adora2b was pharmacologically activated, and this was found to promote axon regeneration and survival of RGCs in adult mice. The magnitude of RGC axon regeneration induced by Adora2b activation was comparable with that after PTEN knockdown, and combining Adora2b activation with PTEN knockdown further enhanced axon regeneration. These findings suggest that targeting Adora2b signaling, either alone or in combination with other pro-regenerative pathways, is likely a strategy for mammalian CNS repair (Wang, 2023).
In the mouse study, DREADDs were used to stimulate the gliotransmitter release. However, the spatiotemporal dynamics of Ca2+ elevations and gliotransmitter release are yet unknown and will be the focus of future studies. Moreover, the current study has focused on mouse retinal glial cells that are positive for GFAP, which is normally found in astrocytes. Future studies are needed to determine whether the stimulation of gliotransmission from other retinal glial types such as Müller glia or microglia could have a similar role in RGC axon regeneration (Wang, 2023).
The molecular and cellular mechanisms underlying complex axon morphogenesis are still poorly understood. This study reports a novel, evolutionary conserved function for the Drosophila Wnk kinase (dWnk) and its mammalian orthologs, WNK1 and 2, in axon branching. This study uncovered that dWnk, together with the neuroprotective factor Nmnat, antagonizes the axon-destabilizing factors D-Sarm and Axundead (Axed) during axon branch growth, revealing a developmental function for these proteins. Overexpression of D-Sarm or Axed results in axon branching defects, which can be blocked by overexpression of dWnk or Nmnat. Surprisingly, Wnk kinases are also required for axon maintenance of adult Drosophila and mouse cortical pyramidal neurons. Requirement of Wnk for axon maintenance is independent of its developmental function. Inactivation of dWnk or mouse Wnk1/2 in mature neurons leads to axon degeneration in the adult brain. Therefore, Wnk kinases are novel signaling components that provide a safeguard function in both developing and adult axons (Izadifar, 2021).
A hallmark in the generation of neuronal cell type diversity is the acquisition of diverse morphologies, which requires the formation of axonal and dendritic compartments ranging from simple to highly complex, depending on the degree of neurite branching. Specifying the degree and pattern of neurite branching is crucial in brain development, as it directly impacts the total number and spatial distribution of synaptic contacts of each circuit element. However, the identity of the molecular effectors determining how diverse, cell-type-specific patterns of axon arborization are established and how they are stabilized as well as maintained throughout the life of an organism remains a major challenge (Izadifar, 2021).
A reverse genetic screen was performed to identify novel regulators of axon branching by utilizing an experimental system in Drosophila that combines efficient single-neuron labeling and simultaneous knockdown of candidate genes. Clear orthologs of selective candidates were then further examined in vertebrates. Using this approach, it was found that loss of the Drosophila dWnk kinase specifically disrupts axon growth and branch patterning of mechanosensory neurons. Surprisingly, unlike other essential regulators of axon branching, this study discovered that dWnk is also continuously required in mature neurons for axon maintenance. Moreover, comparative studies in mouse cortical pyramidal neurons (PNs) provide strong evidence that both of these functions of Wnk kinases are conserved and required in PNs, i.e., long-range projecting mammalian neurons of the central nervous system (CNS) (Izadifar, 2021).
Wnk kinases are present in most multicellular organisms, including plants and some unicellular organisms, but not in yeast. Mammals have four Wnk kinases (WNK1-4) and Drosophila one (dWnk). The WNK ('with no K(lysine)') kinases are catalytically active but are referred to as 'atypical kinases' because a catalytically important lysine residue is swapped from subdomain II to subdomain I. WNK proteins are involved in a broad spectrum of diseases (e.g., hypertension, sensory and autonomic neuropathy, osteoporosis, and many different cancers) (Izadifar, 2021).
The majority of studies on human WNK kinases have been conducted in the context of blood pressure regulation, due to the identification of mutations in human patients with hereditary hypertension (familial hyperkalemia and hypertension [FHHt] or Gordon's syndrome. For this reason, a major focus in dissecting WNK function has been on studying renal regulation of ion transport. However, regulation of ion homeostasis is only one of multiple functions of WNK kinases, and they are broadly expressed, including in the developing as well as mature brain. In rare cases, WNK function has been linked to a severe form of peripheral sensory neuropathy (hereditary sensory and autonomic neuropathy type 2, HSNA2); however, their developmental, cellular, and molecular mechanisms are poorly understood in neurons. The fact, however, that most identified mutations cluster in a neuron-specific alternatively spliced exon (HSN2) of human Wnk1 supports the notion that Wnk1 kinase plays an important role in sensory neurons (Izadifar, 2021).
In the process of studying the role of dWnk kinase in fly sensory neurons, this study identified novel interactors of Wnk kinases. Specifically, it was found that nicotinamide mononucleotide adenylyltransferase (Nmnat), Sarm, and Axundead (Axed) are molecular interactors of dWnk. While Nmnat is broadly required for axon maintenance, Sarm and Axed are primarily studied for their roles as effectors in active axon degeneration (e.g., in Wallerian degeneration) in response to axon injury. This study provides evidence that dWnk and Nmnat have synergistic functions in axon growth and branching but are also required in post-developmental processes to continuously support axon maintenance. The function of dWnk is evolutionarily conserved, as their mouse orthologs WNK1 and WNK2 are both required in cortical PNs during axon morphogenesis as well as maintenance. Genetic epistasis analysis demonstrates that both dWnk and Nmnat functions during axon development and axon maintenance are mediated by antagonizing the axon destruction function of Sarm and Axed. Depletion of axon-protective factors (e.g., dWnk/WNK1/2 and Nmnat) during development leads to axon branching defects, whereas their depletion in mature neurons eliminates their safeguard function and initiates spontaneous axon degeneration in the absence of axon injury (Izadifar, 2021).
This study reports that the function of the Wnk-family kinases (Drosophila dWnk and mammalian WNK1/2) are required in developmental axonal branch patterning as well as axon maintenance. Phenotypic defects observed in mechanosensory axons of dWnk mutant neurons are indistinguishable from defects observed in Nmnat mutant neurons. Both dWnk and Nmnat mutant mechanosensory axons can grow from the periphery to the VNC but require dWnk as well as Nmnat function during axonal branch growth and patterning. Moreover, a post-developmental knockdown of dWnk triggered progressive degeneration in mature and fully developed mechanosensory axons. Such an adult-specific function is also analogous to the well-documented role of Nmnat in axon maintenance. Although Nmnat has been studied most extensively for its role in axon maintenance in injury models or neurodegenerative disease models, it has been reported previously that LOF mutants are lethal and show axonal defects. Furthermore, this study shows that the dual developmental and maintenance functions of dWnk are evolutionary conserved, as knockdown of WNK1 and WNK2 results in remarkably similar axon morphogenesis defects and trigger axon degeneration in mouse cortical neurons. It is concluded that, in neurons, Wnk kinases exert novel functions that are analogous and synergistic to conserved Nmnat functions. The discovery of Wnk kinases having important neuroprotective roles analogous to Nmnat offers new tools and insights to further dissect the complex regulatory network underlying active axon degeneration (Izadifar, 2021).
Formally, the adult maintenance defects that were observed upon Wnk depletion could be a consequence of developmental defects. However, several reasons argue strongly against this possibility: first, the in vivo knockdown of WNK1 and WNK2 after P30 in mouse cortical layer 2/3 PNs triggered degeneration of axons of mature neurons. Second, knockdown of dWnk at post-developmental (late pupal) stages does not alter the axonal branching or targeting of mechanosensory neurons but triggered spontaneous degeneration of adult mechanosensory axons after 3 days post-eclosion. Third, a single-copy RNAi knockdown of dWnk resulted in strong axon branching defects. Fourth, in previous studies, several mutants were identified and characterized that lead to severe axon branching defects of mechanosensory axons. For example, in Dscam1-null mutant clones, the axon branching defects are even more severe than in dWnk mutant clones, yet no sign of axon degeneration was found even in 1 week or older flies. Fifth, even in cases of very short axon branches in dWnk or other mutants with a complete absence of contralateral projecting axon collaterals, the distal part of the mutant axons still reaches the corresponding ipsilateral target area. Given that putative trophic signals would have to support axons on both sides of the VNC, it seems highly unlikely that target-derived trophic signals would only support contralateral projecting axon arbors. In summary, the data provide strong evidence that Wnk kinases have dual roles: first, during developmental axon morphogenesis and, second and independently, during continuous axon maintenance in mature neurons (Izadifar, 2021).
A key question raised by these results is: how similar are the molecular processes in developmental axon growth and branching and adult maintenance? A related question has been discussed in a hallmark review (Raff, 2002). This perspective article discussed the discovery that neurons can activate a self-destructive program independent of general apoptosis. The authors noted that neurons 'apparently have a second, molecularly distinct self-destruct program in their axon.' And the authors raise the incisive question: 'Do neurons also use this second program to prune their axonal tree during development and to conserve resources in response to chronic insults?' (Izadifar, 2021).
It is assumed that 'pruning of axonal branches during development' could-as a cellular mechanism-also be involved in axonal branch patterning as analyzed in this study. Specifically, the developmental axon branching of mechanosensory axons is viewed as a process where continuous competitive interactions among nascent branches select for stabilization or retraction (timescale minutes or few hours). In contrast, the well-characterized pruning of axons during metamorphosis (e.g., mushroom body remodeling) is initiated when axon branches and connectivity have been already established. This type of pruning (remodeling) requires primarily a destabilization of a fully established axon projection and is likely different from axonal branch selection as investigated in this study. Consistent with this idea is the finding that Nmnat is not required in axon pruning of mushroom body neurons (Izadifar, 2021).
The results now provide genetic and molecular data that support the notion that components of a 'distinct self-destruct program' are involved in axon branch stabilization and destabilization. It is further suggested that molecular control mechanisms of axon branching and axon destruction (or preventing axon destruction, i.e., axon maintenance) are mechanistically related. Specifically, the dynamics of axon branching requires the selection of an exuberant number of nascent axon branches by either stabilizing or destabilizing nascent branches. During axon morphogenesis, the majority of filopodia and nascent branches are retracted to maintain just a few that are consolidated into axon collaterals. It seems plausible, therefore, that axon branch retraction in developing neurons might involve molecular effectors that are also required for a distinct type of axon branch pruning. Based on in vitro studies, it is speculated that regulation of de novo protein synthesis could be the molecular process that is targeted in both axon branching and axon maintenance (Izadifar, 2021).
Based on new findings, it is suggested that Wnk as well as Nmnat are necessary components of axon branching as well as axon maintenance. Support for this model comes from the results of genetic epistasis analysis. The loss of dWnk during axon branching is only a problem if Sarm or Axed are present: in the absence of Axed (the most downstream effector of the destruction program in Drosophila known so far), loss of dWnk does not cause defects in axonal branching or maintenance. Implicit to this model is that dWnk is unlikely instructing axon branching but rather provides a safeguard function curbing destructive effectors such that retraction, pruning, or branch destruction can be restricted and constrained in a spatially restricted manner. A further implication of this model is the notion that loss of dWnk effectively represents a gain of function (on switch) of an axonal destruction program in developing axons (Izadifar, 2021).
In this context, it is also interesting to note that expression of Nmnat can compensate (i.e., rescue) for loss of dWnk in both axon branching and maintenance. It is well established that local depletion of Nmnat in adult neurons does directly lead to axon degeneration, i.e., has a role in axon maintenance independent of its developmental role. This is consistent with the new finding that post-developmental inactivation of dWnk or mammalian Wnk1/2 triggers axon degeneration (Izadifar, 2021).
Nmnat has been previously shown to protect from axon degeneration following axotomy by counter-acting Sarm-induced NAD+ depletion and Axed activity. This study shows that, even in the absence of axon injury, loss of Nmnat as well as loss of dWnk or overexpression of dSarm and Axed leads to progressive degeneration of adult mechanosensory axons without injury. This is consistent with previous reports showing that Nmnat loss in sensory neurons of the wing leads to spontaneous axon degeneration in adult flies or that loss of NMNAT2 leads to truncation of peripheral nerve and CNS axon tracts in mice. A role of Wnk1 in axon maintenance is also consistent with the finding that WNK function has been linked to a severe form of peripheral neuropathy (Izadifar, 2021).
The results, therefore, support the notion that both axon morphogenesis and maintenance require a constitutive involvement of Wnk and Nmnat (Izadifar, 2021).
A link between dWnk, Nmnat, and axon destructive factors is further corroborated by biochemical experiments co-expressing these factors and using co-immunoprecipitations to analyze protein-protein interactions of dWnk or mammalian WNK1/2. First, the results suggest the possibility that dWnk, Nmnat, Sarm1, and Axed are able to form mixed complexes. Moreover, mammalian WNK1 can interact with WNK2 and Nmnat2 as well as SARM1. Although previous work has not identified Nmnat proteins as potential Wnk kinase substrates, the current results suggest that this possibility is worthwhile to examine in future experiments. Second, whereas a vertebrate ortholog of Axed has not been described, it was found that mammalian SARM1 overexpression strongly downregulates levels of WNK1, WNK2, NMNAT2, and NMNAT1. However, future experiments will have to confirm that this downregulation of proteins by Sarm1 is also occurring in neurons in vivo (Izadifar, 2021).
It has been reported that inhibition of axon degeneration can be accomplished by increasing or stabilizing levels of Nmnat protein. Moreover, Highwire/Phr1, which is an additional conserved factor functioning in Wallerian degeneration, directly promotes the downregulation of Nmnat, and axon degeneration is strongly inhibited in Highwire/Phr1 mutants. This previously described regulation of Nmnat levels is mediated via mitogen-activated protein kinase (MAPK) signaling and ubiquitin-dependent proteolysis. The findings of this study suggest that Sarm1 may rather inhibit de novo protein synthesis in order to deplete Nmnat and other axon protective factors. Future studies will have to investigate in detail how the SARM1-dependent depletion of NAD+ also leads to a Wnk/Nmnat protein depletion as described in this study. Particularly interesting will be to determine how the destructive activity of Sarm protein can be limited during development to selective axon branch compartments in order to enable local axon branch pruning but prevent progressive axon degeneration (Izadifar, 2021).
Finally, Nmnat function has not only been involved in neuroprotection as a response to injury, such as axotomy, but also in a diverse range of neurodegenerative diseases, such as spinocerebellar ataxia, fronto-temporal dementia (FTD) and Parkinsonism, or glaucomatous optic neuropathy, or following growth factor deprivation. Future studies will need to consider the possibility that the newly identified dWnk and WNK1 and WNK2 kinases may also play similar neuroprotective roles in diverse types of neurodegenerative conditions (Izadifar, 2021).
The crystal structure determination of the armadillo repeat motif (ARM) domain of Drosophila SARM1 (dSARM1(ARM)) is described, that required the combination of a number of sources of phase information in order to obtain interpretable electron-density maps. SARM1 is a central executioner of programmed axon degeneration, a common feature of the early phase of many neurodegenerative diseases. SARM1 is held in the inactive state in healthy axons by its N-terminal auto-inhibitory ARM domain, and is activated to cleave NAD upon injury, triggering subsequent axon degeneration. To characterize the molecular mechanism of SARM1 activation, it was sought to determine the crystal structure of the SARM1 ARM domain. Here, the recombinant production and crystallization of dSARM1(ARM) is described, as well as the unconventional process used for structure determination. Crystals were obtained in the presence of NMN, a precursor of NAD and a potential activator of SARM1, only after in situ proteolysis of the N-terminal 63 residues. After molecular-replacement attempts failed, the crystal structure of dSARM1(ARM) was determined at 1.65Å resolution using the MIRAS phasing technique with autoSHARP, combining data from native, selenomethionine-labelled and bromide-soaked crystals. The structure will further the understanding of SARM1 regulation (Gu, 2021)
Motor neuron axon targeting in the periphery is correlated with the positions of motor neuron inputs in
the CNS, but how these processes are coordinated to form a myotopic map remains poorly understood.
This study shows that the LIM homeodomain factor Islet (Isl)
controls targeting of both axons and dendrites in
Drosophila motor neurons through regulation of the Frazzled (Fra)/DCC receptor. Isl is required for fra
expression in ventrally projecting motor neurons, and isl and fra mutants have
similar axon guidance defects. Single-cell labeling indicates that isl and fra are
also required for dendrite targeting in a subset of motor neurons. Finally, overexpression of Fra
rescues axon and dendrite targeting defects in isl mutants. These results indicate that Fra
acts downstream of Isl in both the periphery and the CNS, demonstrating how a single regulatory
relationship is used in multiple cellular compartments to coordinate neural circuit wiring (Santiago, 2017).
The RP3 motor neurons innervate the NetrinB-expressing muscles 6 and 7 and are enriched for fra mRNA during the late stages of embryonic development, and it was reported previously that, in the absence of fra or Netrin, there are significant defects in the innervation of muscles 6 and 7. This phenotype is also detected in the absence of hb9/exex or isl/tailup, two transcription factors expressed in RP3 as well as in other ventrally projecting motor neurons, suggesting that hb9 or isl may regulate fra. Interestingly, Hb9, Isl, and the LIM homeodomain factor Lim3 were all recently shown to bind directly to the fra locus in vivo, as determined by a genome-wide DNA adenine methyltransferase identification (DAM-ID) analysis performed in Drosophila embryos. However, DAM-ID results do not provide information about the functional significance of the detected binding events or about the cell types in which they occur. To determine whether Hb9, Isl, or Lim3 regulate the expression of fra in embryonic motor neurons, situ hybridization experiments were performed and fra mRNA expression was analyzed with single-cell resolution in embryos mutant for these factors. Only isl is required for fra expression in the RP3 motor neurons at stage 15, when RP axons have reached the ventral muscle field but their final targets have not been selected. 80% of RP3 neurons in abdominal segments A2-A7 in isl/+ embryos are positive for fra mRNA versus 38% in isl mutant embryos. A significant difference was also observed in fra mRNA levels in RP3 neurons between isl mutants and heterozygotes when quantifying pixel intensity from the fra in situ, whereas no difference was detected in the signal of the isl-H-tau-myc transgene. No change was detected in the number or position of RP3 neurons in isl mutants, consistent with previous data demonstrating that Isl is not required for the generation or survival of Drosophila motor neurons. Importantly, no requirement was found for either hb9 or lim3 in regulating fra mRNA expression in any RP motor neurons, demonstrating that isl's effect on fra is specific and could not have been predicted simply from similarities in loss of function phenotypes or from transcription factor binding data (Santiago, 2017).
Hb9 has been shown to be required for robo2expression in RP3. Interestingly, just as hb9 is not required for fra expression in RP neurons, isl is not required for robo2expression. A previous study reported that isl; hb9 double mutants have a stronger intersegmental nerve b (ISNb) phenotype than either single mutant, but muscle 6/7 innervation defects were not quantified. This study scored motor axon guidance defects in isl; hb9 double mutants and found that the double mutants display significantly more muscle 6/7 innervation defects than either single mutant. Similarly, embryos mutant for both robo2and fra have a stronger motor axon phenotype than either robo2or fra single mutants. Note that, because robo2, fra double mutants have severe defects in midline crossing, motor axon phenotypes should be interpreted with caution . These results show that Hb9 and Isl act in parallel to regulate distinct downstream programs in RP3 neurons, demonstrating how combinations of transcription factors result in specific cell surface receptor profiles and axon trajectories (Santiago, 2017).
To determine whether isl and fra act in the same genetic pathway during RP3 guidance, embryos mutant for both genes were examined. In isl-null mutants, 20% of hemisegments lack muscle 6/7 innervation, whereas fra-null mutants have a significantly stronger phenotype (34% of hemisegments). isl, fra double mutants do not have more muscle 6/7 innervation defects than fra single mutants, consistent with isl and fra acting in the same pathway. If fra acts downstream of Isl during motor axon targeting, then it was reasoned that restoring Fra expression in isl mutant neurons might rescue muscle 6/7 innervation. Indeed, it was found that pan-neural overexpression of Fra in isl mutants partially but significantly rescues these defects. The difference between genotypes was most noticeable when hemisegments were counted in which a growth cone stalls at the 6/7 cleft as well as those in which it fails to reach it. In isl mutants, a growth cone stalls at or fails to reach the 6/7 cleft in 27% of hemisegments compared with 15% of hemisegments in sibling mutants overexpressing Fra. The data was also analyzed by comparing the number of embryos with 6/7 innervation defects. In isl mutants, 0% of embryos have no 6/7 innervation defects in A2-A6, 44% have one defect, and 56% have two or more defects. In contrast, in isl mutants overexpressing Fra, 29% of embryos have no innervation defects, 29% have one defect, and 41% have two or more defects. The incomplete rescue could be due to differences in the timing or levels of GAL4/UAS-mediated expression of Fra compared with its endogenous regulation or could indicate that Isl regulates additional downstream effectors important in this process. Nevertheless, these data strongly suggest that Fra is an essential downstream effector of Isl during the guidance of the RP3 axon to its target muscles (Santiago, 2017).
To further investigate the relationship between isl and fra, whether ectopic expression of isl is sufficient to induce fra expression was tested. These experiments used the apterous (ap) neurons. The axons from this subset of interneurons form a single fascicle on either side of the midline that are labeled by ap-Gal4. The ap neurons express low levels of fra, do not express isl, and do not cross the midline. Fra overexpression causes ectopic midline crossing of ap axons. Overexpression of Isl with ap-Gal4 produced high levels of midline crossing, phenocopying the effect of Fra overexpression. In stage 17 control embryos, ap axons cross the midline in 12% of segments, whereas in embryos overexpressing UAS-Isl with ap-Gal4, ap axons cross the midline in 60% of segments. This phenotype is dose-dependent because embryos with two copies of an UAS-Isl insertion display significantly more ectopic midline crossing than embryos with one insert (Santiago, 2017).
To determine whether Isl overexpression results in fra induction, the expression of fra mRNA in situ was examined in ap neurons. In stage 15 wild-type embryos, a low percentage of ap neurons express fra (25% of ventral ap clusters were fra+). In contrast, in embryos overexpressing isl from two UAS-Isl inserts, 37% of the ventral ap clusters were fra+. To test whether the ectopic crossing phenotype depends on fra function, Isl was overexpressed in embryos homozygous for a null allele of fra. Strikingly, removing fra completely suppresses the crossing phenotype, indicating that fra is required for Isl to produce its gain-of-function effect. Although it cannot be ruled out that Isl affects the expression of other genes in the Fra pathway to cause midline crossing, these results demonstrate that ectopically expressing Isl causes an increase in fra expression and a fra-dependent axon guidance phenotype and suggest that the functional relationship between isl and fra may be used in multiple contexts (Santiago, 2017).
Fra mutants have defects in RP axon midline crossing, as shown by retrograde labeling of single motor neurons. In addition, Netrin-Fra signaling controls the medio-lateral position of dendrites in several groups of motor neurons. Therefore, it was asked whether isl regulates midline crossing or RP3 dendrite development through fra. A genetic strategy was used to label single motor neurons by mosaic expression of a membrane-tethered GFP transgene under the control of lim3b-GAL4, which labels RP motor neurons, sensory neurons, and several other motor and interneuron populations. RP3 neurons were identified by the stereotyped position of the RP3 cell body and by the targeting of its axon to muscles 6 and 7. Because of the axon targeting defects observed in isl and fra mutants, cell body position was used to identify RP3 neurons in mutants. By this approach, significant midline crossing defects were detected in RP3 axons in fra mutants. Surprisingly, however, no defects were observed in RP axon midline crossing in isl mutants (Santiago, 2017).
Isl and fra expression both initiate earlier than stage 13, the time at which RP axons cross the midline. Therefore, whether isl is required for fra expression was examined during the early stages of commissural axon guidance. Interestingly, isl was not required for fra expression at stage 13 in any of the ventrally projecting RPs. In contrast, in stage 15 isl mutant embryos from the same collection, a decrease in was observed fra expression in RP1 and RP3. The temporal pattern of fra expression in RP motor neurons is dynamic, so that a larger proportion of RP1 and RP3 neurons express fra mRNA during late embryogenesis than during the stages of midline crossing. A requirement was detected for isl in regulating fra in RP1 and RP3 as early as stage 14, when the RP motor axons have exited the CNS. Taken together, these results suggest that isl is not essential for early fra expression but required for fra expression during the late stages of motor neuron differentiation. The stages at which a requirement was detected for isl in regulating fra correspond to when RP3 axons are exploring their ventral muscle targets, consistent with a model in which Isl instructs the final stages of RP3 axon targeting through Fra (Santiago, 2017).
Another essential feature of Drosophila larval motor neurons that is established late in embryogenesis is the morphogenesis and targeting of their dendrites in the ventral nerve cord. Motor neuron dendrites begin to form as extensions off the primary neurite at stage 15, a stage when a requirement is detected for isl in regulating fra. By early stage 17 (15 hr after egg laying, AEL), RP3 has assumed its stereotyped morphology, consisting of a small ipsilateral projection extending from the soma and a large dendritic arbor forming off the contralateral primary neurite (Santiago, 2017).
The FLP-out genetic labeling strategy was used to visualize individual late-stage RP motor neurons and analyze their dendrites. Focus was placed on the large contralateral arbor of the RP motor neurons that spans one side of the nerve cord in wild-type embryos and forms branches that extend into several medio-lateral zones. Analyses using isl-tau-myc and lim3a-tau-myc transgenes confirmed that the RP cell bodies retain their stereotyped positions in isl mutants and that the relative dorsal-ventral positions of RPs 1/4, 3, and 5 are preserved, allowing identification of distinct classes of RP motor neurons (Santiago, 2017).
Most RP3 neurons in late-stage isl/+ embryos neurons form contralateral dendritic arbors that send projections into the zone between the medial FasII+ axon pathways and the intermediate FasII+ pathways, hereafter referred to as the 'intermediate zone,' consistent with previously published images of RP3 neurons from wild-type embryos. Interestingly, the dendritic morphology of RP3 was distinct from that of a related neuron, RP5, that also expresses Isl and Lim3b-Gal4 and that can be unambiguously identified in both wild-type and mutant embryos because its cell body is found in a more ventral position than the other RP neurons. In wild-type embryos, the RP5 axon targets muscles 12 and 13 (VL1 and VL2) as well as other ventral muscles. Most RP5 neurons in isl/+ embryos exclusively target their dendrites to the lateral zone of the neuropile. Furthermore, the difference observed in the dendritic targeting of RP3 and RP5 neurons correlates with a difference in fra expression. Although fra expression in RP3 and RP5 neurons in control embryos is comparable when RP axons are crossing the midline, by stage 15, significantly fewer RP5 than RP3 neurons express fra. Interestingly, isl is not required for the low levels of fra expression in late-stage RP5 neurons, in contrast to its role in promoting high levels of fra in late-stage RP3 neurons (Santiago, 2017).
Finally, endogenous Netrin expression was monitored in late-stage nerve cords using a Myc-tagged NetB knockin allele, and significant enrichment of Netrin protein was detected in the area between the intermediate and medial FasII+ axon bundles. This area corresponds to the zone where contralateral dendritic projections from RP3 neurons were detected, suggesting that high levels of Fra in RP3 may instruct the formation of dendritic arbors in this region in response to Netrin (Santiago, 2017).
RP motor neuron dendrites were examined in isl mutant embryos to determine whether Isl regulates dendritic position or morphogenesis through Fra or other effectors. No significant difference in the morphology or medio-lateral position of RP5 dendrites was observed between heterozygous and mutant embryos. In striking contrast, many RP3 neurons in isl mutants fail to extend contralateral dendrites into the intermediate zone. Instead, the dendrites of these RP3 neurons remain fasciculated with the intermediate FasII+ axon pathways and do not send medial extensions toward the midline. To more quantitatively measure medio-lateral position and to address the possibility that defects in targeting are secondary to defects in outgrowth, RP3 neurons were traced using Imaris software and total contralateral dendrite lengths and the total number of dendrite tips were measured. Total length of contralateral dendrites was also measured in the intermediate zone of the neuropile, defined as the area between the medial FasII+ and the intermediate FasII+ axon pathways. Although RP3 neurons displayed increased variability in the size of their dendritic arbors in isl mutants, there was no significant difference in the total length or tip number of RP3 dendrites between isl mutants and heterozygotes, suggesting that targeting defects in isl mutants are not likely due to reduced outgrowth. However, the ratio of RP3 dendrites in the intermediate zone over total RP3 dendrite length was significantly reduced in isl mutants, confirming that isl mutant RP3 dendrites are shifted laterally relative to controls (Santiago, 2017).
The dendrites of RP3 neurons were examined in fra/+ and fra mutant embryos. As with isl mutants, cell body position was used to identify RP3 neurons, and neurons with ambiguous positions were excluded. In fra mutant RP3 neurons whose axons fail to cross the midline, a single dendritic arbor forms off the ipsilateral primary neurite, and this arbor was traced. A significant lateral shift was observed in the position of RP3 dendrites in fra mutants both by scoring for the presence of dendrites in the intermediate zone and by quantitative analysis of the dendrites of traced neurons. The lateral shift in fra mutants was more pronounced than in isl mutants, consistent with the observation that some RP3 neurons retain fra expression in the absence of isl. Of note, the lateral shift phenotype did not correlate with whether the RP3 axon had crossed the midline because it was detected at similar frequencies in both contralateral and ipsilateral arbors. Curiously, several RP3 contralateral dendritic arbors appeared reduced in size in fra mutants, whereas this phenotype was not seen in control embryos. However, as in isl mutants, there was no significant change in the total dendrite length or tip number in fra mutants compared with their sibling controls, although there was increased variability in the sizes of dendritic arbors in the mutants. These findings are consistent with previous reports that Netrin-Fra signaling does not play a major role in regulating the outgrowth of motor neuron dendrites in the nerve cord (Santiago, 2017).
A single-cell labeling method allows precise description of the axon targeting defects in isl and fra mutants and determine whether they correlate with defects in dendrite position. Axon and dendrite targeting occur at approximately the same developmental stage, and there is no evidence that one process depends on the other. Importantly, previous studies using retrograde labeling of motor neurons in mutant embryos were not able to address this question because they relied upon motor axons reaching the correct muscles to be visualized (Santiago, 2017).
To determine whether defects in dendrite position correlate with defects in axon targeting, both phenotypes were scored in single labeled RP3 neurons in embryos with muscles fully preserved following dissection. All of the RP3 axons that could be scored in isl heterozygous embryos innervated the muscle 6/7 cleft . In contrast, 18 of 26 isl mutant RP3 axons innervated muscles 6/7, and eight stalled at the 6/7 cleft or earlier along RP3's trajectory or bypassed the choice point. In fra mutant embryos, 10 of 22 RP3 neurons failed to innervate the muscle 6/7 cleft and stalled at or bypassed the choice point. This phenotype is stronger than the frequency at which a complete loss of muscle 6/7 innervation in isl or fra mutants was detected by scoring with anti-FasII. To determine whether this enhancement was due to the heat shock (H.S.) step that is required for genetic labeling, defects were scored using anti-FasII in embryos heat-shocked for either 5 min or 1 hr; it was found that the 1-hr H.S. mildly enhances muscle innervation defects in isl mutants (to 30.4%) whereas a 5-min H.S. does not (to 24.7%, data not shown). Importantly, the two H.S. protocols did not result in any difference in the frequency of dendrite targeting defects observed in isl mutants because 7 of 17 RP3 dendrites in isl mutants are shifted laterally in embryos treated with 1-hr H.S, and 9 of 16 dendrites are shifted after 5-min H.S (Santiago, 2017).
Surprisingly, no correlation was detected between axon and dendrite defects in isl mutants. Although 5 of 26 RP3 neurons displayed defects in both axons and dendrites in isl mutants, 12 of 26 neurons showed defects in one process but not the other). A similar analysis in fra mutants revealed that 8 of 22 RP3 neurons displayed defects in both muscle 6/7 innervation and dendrite position, whereas 8 of 22 displayed normal targeting in one process but not the other. These data suggest that axon and dendrite targeting can occur independently within an individual RP3 neuron and that the central targeting defects observed in isl mutants are not likely to be secondary to defects in muscle innervation (Santiago, 2017).
It was next asked whether isl and fra regulate dendrite development in other classes of motor neurons. RP1 and RP4 also express isl, fra, and lim3b-Gal4. A requirement was detected for isl in regulating fra expression in RP1, but not in RP4, at stage 15. Interestingly, most RP1 neurons, like RP3 neurons, retain high levels of fra at this stage, whereas few RP4 neurons express fra in late-stage control embryos. Previous descriptions of RP1 and RP4 neurons indicate that they form contralateral dendritic arbors of distinct morphologies; RP1's dendritic arbor is taller and found more medially. However, because the axons of RP1 and RP4 target adjacent muscles external to muscles 6 and 13, and their cell bodies are found close to the midline at a similar dorsal-ventral position, they could not nr unambiguously distinguished in single-cell labeling experiments. Nevertheless, when RP1 and RP4 neurons were scored together, a significant lateral shift was observed in the position of RP1 and RP4 dendrites in isl mutants compared with heterozygous siblings: 3 of 22 RP1 and RP4 dendritic arbors were excluded from the intermediate zone in isl heterozygous embryos (14%) compared with 16 of 22 in mutant embryos. A similar phenotype was detected in RP1 and RP4 dendrites in fra mutants. Specifically, 6 of 24 RP1 and RP4 dendritic arbors were excluded from the intermediate zone in heterozygotes (25%) compared with 17/19 in fra mutants. Although additional work will be necessary to determine whether the defects in dendrite position that were detect in RP1 and RP4 neurons in isl mutants correlate with changes in fra expression, these data demonstrate that Isl is required for high levels of fra expression in at least two classes of motor neurons (RP1 and RP3), both of which require isl and fra for dendritic targeting (Santiago, 2017).
To directly test whether isl regulates RP3 dendrite position through its effect on fra expression, a UAS-HA-Fra transgene was overexpressed using lim3b-GAL4 in isl mutants and the hsFLP technique was used to sparsely label RP motor neurons. Strikingly, in isl mutants overexpressing Fra, 0 of 21 RP3 contralateral dendritic arbors were excluded from the intermediate zone compared with 8 of 22 (36%) in sibling mutants lacking the UAS-Fra transgene. To quantitatively measure dendrite position, traces of RP3 dendrites were obtained. A robust rescue of the lateral shift phenotype was detected in isl mutants, as measured by the length of dendrites in the intermediate zone over the total dendrite length. Indeed, the ratio of dendrites in the intermediate zone in rescued mutants was higher than in heterozygous controls, perhaps reflecting a gain-of-function effect caused by artificially high levels of Fra from transgenic overexpression. Importantly, Fra overexpression did not have any effect on total dendritic arbor lengths or tip numbers, strongly arguing that the rescue that was observe is not caused by an increase in the total size of the arbors. Although it cannot be ruled out that Isl regulates dendrite position in part through additional effectors, the observation that cell-type-specific overexpression of Fra in isl mutants rescues dendrite targeting provides compelling support for the model that fra acts downstream of isl to control RP3 dendrite morphogenesis. Together with the demonstration that isl directs RP3 motor axon targeting through the regulation of fra, it is concluded that isl coordinately regulates the targeting of axons in the periphery and of dendrites in the CNS through a common downstream effector (Santiago, 2017).
In the vertebrate spinal cord, the position of motor neuron cell bodies correlates with the targeting of their axons in the periphery (Catela, 2015). This myotopic map may be established through the action of transcription factors that coordinately control cell migration and axon guidance. In particular, Lhx1 and Isl1 are expressed in limb-innervating lateral motor column (LMC) motor neurons and regulate the trajectory of their axons as well as the medio-lateral settling position of their cell bodies . Lhx1 and Isl1 regulate axon guidance through EphA4 and EphB receptors, respectively, and a recent study suggests that Lhx1 regulates cell body position through a distinct effector, the Reelin signaling protein Dab-1 (Santiago, 2017).
In Drosophila, unlike in vertebrates, the position of motor neuron cell bodies does not necessarily correlate with the targeting of their axons in the periphery because neurons that innervate adjacent muscles can be found far apart within a segment. Instead, recent studies have shown that both the larval and the adult Drosophila nervous systems use a myotopic map in which the position of motor neuron dendrites, rather than their cell bodies, correlates with the position of their target muscles (Brierley, 2009, Mauss, 2009). This may be a conserved feature of motor systems across phyla because the dendritic patterning of at least four motor neuron pools in the spinal cord correlates with muscle target identity in the mouse (Santiago, 2017).
Slit-Robo, Netrin-Fra, and Sema-Plexin signaling have been shown to control motor neuron dendrite targeting in Drosophila, and rescue experiments suggest that these guidance receptors act cell-autonomously in this process (Brierley, 2009, Mauss, 2009, Syed, 2016). In addition, the initial targeting of motor neuron dendrites in the embryo is largely unaffected by manipulations that affect the position or the activity of pre-synaptic axons or the presence of muscles, suggesting that this process is likely under the control of cell-autonomous factors, although these remain unidentified (Santiago, 2017).
This study has addressed several key questions about how motor neuron dendrite targeting is specified in Drosophila. First, it was shown that fra expression in two classes of motor neurons (RP3 and RP5) correlates with the medio-lateral position of their dendrites. Previous studies suggested that different classes of motor neurons express different levels of guidance receptors to direct the position of their dendrites, but this has not been demonstrated. Second, Isl, which was previously shown to regulate axon targeting in a subset-specific way, was also shown to regulate dendrite targeting. Third, it was found that Isl regulates both processes through fra. Surprisingly, no correlation was found between axon and dendrite phenotypes in isl mutants. The absence of a correlation suggests that the dendrite positioning defects are not secondary to defects in target selection, consistent with a previous study in which the general patterning of motor neuron dendrites was not disrupted in muscle-less embryos. However, additional experiments that disrupt axon targeting and monitor the medio-lateral position of dendrites will be necessary to confirm that the two occur independently (Santiago, 2017).
Future work will also be necessary to identify additional transcription factors that specify motor neuron dendrite development. A role has been identified for Hb9 in regulating robo2and robo3 expression, but it is not known whether these receptors regulate motor neuron dendrite development. No change was detected in robo1 mRNA levels in RP3 neurons in either hb9 or isl mutants. Robo signaling could be regulated post-transcriptionally. Comm is required for midline crossing of motor neuron dendrites and may endogenously regulate their medio-lateral position. The temporal pattern of comm expression does not support a role in dendrite targeting, however, because comm is not expressed in RP motor neurons at late stages of embryogenesis (Santiago, 2017).
The functional consequences of dendrite targeting defects remain to be explored. It is likely that shifting the position of motor neuron dendrites alters their connectivity, but testing this hypothesis will require identifying the pre-synaptic neurons that impinge on the RPs during locomotive behavior. Forcing a shift in the position of dendrites of dorsally projecting motor neurons does not abolish their connectivity with known pre-synaptic partners but does change the number of contacts established. In mice, the ETS factor Pea3/Etv4 is required for the dendritic patterning of a subset of motor neurons, and electrophysiological recordings reveal changes in connectivity in Pea3 mutant spinal cords. It will be of high interest to investigate whether analogous defects are detected in isl or fra mutant embryos (Santiago, 2017).
Drosophila Isl was initially described as a subset-specific regulator of axon guidance. More recently, Wolfram (2012) demonstrated that Isl also acts instructively to establish the electrophysiological properties of RP motor neurons through repression of the potassium ion channel Shaker. The curreng data show that, in addition to regulating the axonal trajectory and the electrophysiological properties of the RP3 neuron, Isl also establishes its dendritic position. Terminal selectors have been defined as transcription factors that coordinately regulate gene programs conferring multiple aspects of a neuron's identity, including its neurotransmitter phenotype, ion channel profile, and connectivity. Unlike the early-acting factors that function transiently to specify cell fate, terminal selectors are expressed throughout the life of an animal and are required for the maintenance of neural identity. Although there are several examples of transcription factors that act this way, it remains unclear how widespread a phenomenon it is. Does Isl fit the criteria for a terminal selector? Isl is not required for all aspects of RP3 identity because RP neurons retain expression of other motor neuron transcription factors in isl mutants, and their axons exit the nerve cord. Future work will be necessary to determine whether Isl is required throughout larval life for the maintenance of RP3's physiological and morphological features and to what extent Isl coordinately establishes multiple features of RP neuron identity (Santiago, 2017).
Co-expressed transcription factors could act synergistically to regulate specific downstream programs, in parallel through completely distinct effectors, or by some combination of the two mechanisms. Indeed, examples of all of these scenarios have been described. Both in vitro and in vivo studies demonstrate that, in vertebrate spinal motor neurons, Isl1 forms a complex with Lhx3 and that the Isl1-Lhx3 complex binds to and regulates different genes than Lhx3 alone or than a complex composed of Isl1 and Phox2b, a factor expressed in hindbrain motor neurons. In a subset of spinal commissural neurons, Lhx2 and Lhx9 act in parallel to promote midline crossing through upregulation of Rig-1/Robo3. In Drosophila dorsally projecting motor neurons, Eve, Zfh1, and Grain act in parallel to promote the expression of unc5, beat1a, and fas2, although Eve also regulates additional targets important for axon guidance that are not shared by Zfh1 or Grain (Santiago, 2017).
This study shows that Isl and Hb9 act in parallel through at least two distinct effectors and proposes that they regulate their targets by different mechanisms. Hb9 likely indirectly promotes robo2 expression by repressing one or multiple intermediate targets because its conserved Engrailed homology repressor domain is required for its function in motor axon guidance and for robo2 regulation. In vertebrate motor neurons, Isl1 forms a complex with Lhx3 to directly activate several of its known targets. A recent genome-wide DAM-ID analysis found that Isl binds to multiple regions within and near the fra locus in Drosophila embryos, suggesting that it may directly activate fra. The finding that lim3 is not required for fra expression in RP motor neurons, together with evidence that Isl can alter the electrical properties of muscle cells independently of Lim3, suggest that Drosophila Isl does not need to form a complex with Lim3 for all of its functions. Future research will be necessary to detect Isl binding events in embryonic motor neurons, although these experiments are challenging when binding occurs transiently or in a small number of cells. Interestingly, overexpression of Isl using ap-Gal4 or hb9-Gal4 induces fra only in certain subsets of these neurons, consistent with a model in which Isl binds to the fra locus in a cell-type-specific manner. The generation of many large-scale datasets for transcription factor binding sites presents the field with the task of reconciling these data with clearly defined genetic relationships during specific biological processes. This study and others have initiated this effort, but it will be important to investigate the functional significance of other putative transcription factor-effector relationships to achieve a better understanding of how transcriptional regulators control cell fate (Santiago, 2017).
Axon degeneration is a hallmark of neurodegenerative disease and neural injury. Axotomy activates an intrinsic pro-degenerative axon death signaling cascade involving loss of the NAD+ biosynthetic enzyme Nmnat/Nmnat2 in axons, activation of dSarm/Sarm1, and subsequent Sarm-dependent depletion of NAD+. This study has identified (Axed) as a mediator of axon death. axed mutants suppress axon death in several types of axons for the lifespan of the fly and block the pro-degenerative effects of activated dSarm in vivo. Neurodegeneration induced by loss of the sole fly Nmnat ortholog is also fully blocked by axed, but not dsarm, mutants. Thus, pro-degenerative pathways activated by dSarm signaling or Nmnat elimination ultimately converge on Axed. Remarkably, severed axons morphologically preserved by axon death pathway mutations remain integrated in circuits and able to elicit complex behaviors after stimulation, indicating that blockade of axon death signaling results in long-term functional preservation of axons (Neukomm, 2017).
Maintenance of the morphological integrity of neurons is essential for sustained nervous system function throughout an animal's lifespan. Nervous system injury or neurological disease leads to axonal and synaptic degeneration and, in turn, loss of neural circuit connectivity and function. Molecular pathways driving axonal degeneration remain poorly defined in any context; however, recent work on Wallerian degeneration (WD) has revealed that axon injury activates an intrinsic, conserved, pro-degenerative (axon death) signaling pathway. Previously identified dSarm/Sarm1 (sterile α/Armadillo/Toll-Interleukin receptor homology domain protein) as a key mediator of axon death signaling. Loss of dSarm in Drosophila, or Sarm1 in mouse, resulted in severed distal axons remaining morphologically preserved for weeks after injury (Gerdts, 2013, Osterloh, 2012), indicating that dSarm/Sarm1 pro-degenerative signaling is an ancient mechanism used by axons to drive self-destruction. How dSarm/Sarm1 signals to execute axon death remains unclear, but dSarm/Sarm1 has recently been linked to the NAD+ metabolic pathway, which based on extensive evidence appears to be a central mediator of axonal integrity (Neukomm, 2017).
The first evidence supporting a role for NAD+ in axon maintenance came from the identification and characterization of the slow Wallerian degeneration (WldS) mouse, where severed axons fibers survived for weeks when detached from their cell bodies. This remarkable neuroprotective phenotype effect was due to a chromosomal rearrangement that led to the generation of the novel WldS molecule, a fusion protein consisting of the NAD+ biosynthetic enzyme nicotinamide mononucleotide adenylyltransferase 1 (Nmnat1) and a short fragment of Ube4b. Consistent with a positive role for NAD+ in sustaining axons, NAD+ levels were found to plummet in axons immediately prior to granular fragmentation. Axon degeneration could be rescued by exogenous NAD+ or its precursors, and axotomy-induced NAD+ depletion was blocked by WldS. Numerous studies have demonstrated neuroprotective roles for NAD+-related metabolites. The current proposed mechanism for activation of axon degeneration after injury is the depletion of Nmnat2, a labile form of Nmnat found in mammalian axons that is normally transported down axons from the soma. Nmnat2 is seen as a critical regulator of axon survival: its half-life approximates the latent phase prior to explosive axon fragmentation, depletion of Nmnat2 from axons induces spontaneous degeneration, and stabilization of Nmnat2 can phenocopy the effects of WldS (Neukomm, 2017).
The second line of evidence supporting a role for NAD+ in axonal protection came from drug screens for molecules that promoted neurogenesis (or neuroprotection) in vivo. P7C3 was identified as an activity-enhancing compound of nicotinamide phosphoribosyltransferase (Nampt), a rate-limiting enzyme in the NAD+ salvage pathway, which likely leads to increased levels of NAD+ in injured axons to help sustain integrity. Derivatives of the P7C3 series have since been shown to be neuroprotective in many models of neurodegenerative disease, and in some models of neural injury . However, whether P7C3 harbors the ability to attenuate axon death remains to be determined (Neukomm, 2017).
Finally, recent work has demonstrated that NAD+ depletion after axotomy is blocked in Sarm1-/- mutants, and dimerization of the Sarm1 TIR domain can drive rapid depletion of NAD+ from cells and axons (Gerdts, 2015). Surprisingly, it appears that the Sarm1 TIR domain harbors endogenous NAD+ hydrolase activity (Essuman, 2017). Thus an emerging stepwise model of WD is that axotomy leads to depletion of labile pools of Nmnat2 in axons detached from their cell bodies; this in turn results in an initial depletion of NAD+ levels due to lack of new NAD+ synthesis and/or salvage, dSarm/Sarm1 signaling is then activated followed by rapid depletion of NAD+ levels below a threshold needed to maintain axonal integrity, and finally explosive degeneration of the axon ensues.
NAD+ depletion from axons is a compelling model for WD, although alternative mechanisms cannot be excluded, due to limited understanding of the genetics of axon death signaling. Besides dSarm/Sarm1, only one other molecule, the E3 ubiquitin ligase Highwire/Phr1, has been shown to be required for axon death in vivo: loss-of-function mutations in highwire/Phr1 potently suppress axon death in fly nerve injury models and mammalian sciatic nerve lesion experiments. Highwire/Phr1 likely acts upstream of dSarm/Sarm1, and is required for the normal turnover of Nmnat/Nmnat2. Loss of Highwire/Phr1 is therefore proposed to stabilize Nmnat/Nmnat2 and maintain axonal pools of NAD+. Are there other signaling molecules acting downstream of dSarm/Sarm1 to execute axonal death, or is NAD+ depletion the final step? Sarm1 has also been reported to drive axon death through a downstream MAPK signaling cascade (Yang, 2015), similar to the signaling mechanism used by C. elegans TIR-1 during regulation of odorant receptor expression; however, this remains controversial. A recent contradictory study argues that MAPKs exert their effect by fine-tuning levels of Nmnat2 upstream of Sarm1 signaling (Neukomm, 2017).
This paper reports the identification and characterization of Axundead (Axed), an axonal BTB and BACK domain protein that signals downstream of dSarm. Axed is essential for injury-induced axon death signaling, and axed mutations can fully suppress degeneration induced by activated dSarm, or complete elimination of Nmnat activity from axons. Thus, Axed is a novel pro-degenerative signaling molecule, and the neuroprotective effects of loss of Axed exceed those of dsarm null mutants (Neukomm, 2017).
This study presents the identification and initial characterization of the BTB and BACK domain molecule Axed. Loss of Axed function was sufficient to block axon death for the lifespan of the fly, and axed mutants were neuroprotective in all neurons tested. Axed, like dSarm, appears to function selectively in axon death during WD, as axed mutants blocked neither cell death nor developmental pruning of axons or dendrites, supporting the notion that the Nmnat-depletion/dSarm/Axed signaling pathway is engaged specifically in response to axonal injury. However, it remains possible that while Axed or dSarm elimination is not sufficient to block cell death, redundant genetic pathways might work together with dSarm and Axed in the context of apoptotic cell death (Neukomm, 2017).
Axed encodes a previously uncharacterized BTB and BACK domain protein. The best-characterized role for BTB-containing proteins is the recruitment of substrates to
Cullin Ring Ubiquitin Ligase (CRL) complexes for ubiquitin tagging and proteasome degradation, where BTB domains function in homo- or hetero-dimerization while the BACK domain (often with the BTB domain) interacts directly with Cullins. Typically, the C-terminal regions in BTB domains molecules function within CRL complexes to bind target substrates designated either for ubiquitination by the CRL and subsequent degradation by the proteasome or for signaling. The BTB and BACK domains of Axed are required for optimal Axed function in vivo based on their partial abilities to rescue axon death phenotypes in axed mutant backgrounds. In contrast, the C terminus appears to be absolutely essential for Axed function as AxedδCterm completely fails to rescue axon death. This is not likely due to destabilization of the molecule lacking the C terminus, since AxedδCterm can be stably expressed in Drosophila S2 cells. Despite extensive attempts to implicate the wide array of CRLs in Drosophila in axon death and Axed function, no evidence was found supporting a role for CRLs in axon degeneration. Given that there are five Cullin-like molecules in the Drosophila genome, it seems possible that genetic redundancy between Cullins might explain the lack of phenotype. Alternatively, Axed could be functioning to promote axon death in a completely novel Cullin-independent manner. Future studies aimed at identifying direct binding partners for Axed will be essential to resolve these issues. If Axed functions as a bridging molecule for CRL ubiquitination activity, of particular interest will be identifying molecules that bind to the Axed C-terminal putative substrate binding domain (Neukomm, 2017).
Sarm1 functions in axons after injury (Gerdts, 2015). Given that Axed functions genetically downstream of Sarm1, it is proposed that Axed also functions in axons. Using a functional, endogenously enhanced GFP-tagged version of Axed (AxedeGFP), it was found that Axed protein is enriched in the neuropil in both the larva and the adult nervous system. The Drosophila larval neuropil is highly enriched for dendrites and axons and is the site of all CNS synapse formation. AxedeGFP signals overlapped extensively with axons, dendrites, and synapses, further supporting a cell-autonomous role for Axed in neurons during axon death. AxedeGFP is also present in the adult neuropil, and AxedeGFP signals transiently increased at 4 and 6 hr in the antennal lobe after injury but returned to baseline levels by 24 hr, a time point at which most severed axons have fragmented. The return to baseline levels likely represents AxedeGFP staining in local and projection interneurons in the antennal lobe that were not severed by antennal ablation. Why AxedeGFP signals might transiently increase remains unclear. Perhaps AxedeGFP is relocalized in a way that increases GFP exposure, or new AxedeGFP protein may be locally synthesized. Regardless of the precise mechanisms, the observations indicate that AxedeGFP dynamics in vivo are sensitive to axonal injury (Neukomm, 2017).
axed mutant phenotypes are indistinguishable from dsarm mutants with respect to axon preservation after injury. In addition, axed mutants completely blocked the pro-degenerative activity of the gain-of-function dSarm molecule dSarmΔdsarm in vivo. These genetic data strongly support a model whereby Axed functions genetically downstream of dSarm, and argue that dSarm and Axed drive axon death through the same genetic pathway (Neukomm, 2017).
dSarmΔdsarm appears to induce a Wallerian-like program in vivo based on several observations. First, the explosive fragmentation observed is morphologically similar to WD. Second, dSarmΔdsarm-induced degeneration can be suppressed partially by WldS, whose activity is highly selective to WD. Third, dSarmΔdsarm pro-degenerative activity can be fully suppressed in axed mutants. Finally, all structural and biochemical studies to date support the notion that elimination of the N-terminal ARM domain of dSarm (Drosophila), TIR-1(C. elegans), or Sarm1 (mammals) leads to the production of a gain-of-function molecule that activates signaling. That dSarmΔdsarm signaling in the absence of injury is potently suppressed by axed mutants provides a compelling argument for Axed to act downstream of dSarm. The nature of the genetic program that drives death of neuronal cell bodies after dSarm/Sarm1 activation remains mysterious. It could not be blocked with the broad caspase inhibitor P35. Related studies in mouse found a lack of evidence for apoptotic signaling molecules, necroptosis, and parthanatos in death induced by activated Sarm1. The observation that it was possible to induce robust degeneration of axons, dendrites, and cell bodies by adult-specific induction of dSarmΔdsarm in adult PDF+ neurons, even in the absence of injury, suggests that dSarmΔdsarm could provide a useful tool for conditional removal of selected neurons from adult circuits for functional studies (Neukomm, 2017).
In the larger context of axon death signaling, the finding that mutations in hiw do not suppress dSarmΔdsarm in vivo agrees with the proposed model that Hiw/Phr1 acts genetically upstream of dSarm/Sarm1 and Nmnat degradation. The position of the MAPK signaling cascade relative to dSarm/Sarm1 is still debated in the field and remains to be determined. The fact that the Drosophila genome houses only a single JNK family member, and the observation that null alleles had no axon death phenotype, argues strongly against a central role for JNK signaling in axon death (Neukomm, 2017).
With respect to their roles in the functional disassembly of axons, this study has demonstrated that blocking axon death with mutations in dsarm, axed, or highwire is sufficient to preserve axons in a functional state in neural circuits where they can elicit complex grooming behaviors for weeks after axotomy. Therefore, axon death mutants, like the WldS molecule, exert their neuroprotective effects very early in the axon death genetic program, and their therapeutic blockade is likely to lead to the preservation of functional axons in the context of neurological disease (Neukomm, 2017).
A number of recent studies have led to the following model for axon death signaling: (1) axotomy results in the degradation of pools of labile Nmnat/Nmnat2 in distal severed axons; (2) depletion of Nmnat/Nmnat2 in turn leads to decreases in axonal NAD+; (3) loss of Nmnat/Nmnat2 or decreased NAD+ somehow activates dSarm/Sarm1 signaling, and (4) Sarm1 NAD+ hydrolase activity then drives pathological depletion of axonal NAD+ pools, causing catastrophic energy failure that ultimately drives axon degeneration. A number of observations were made that are difficult to rectify with a simple Nmnat2/Sarm1-dependent NAD+ depletion model. The first is that expression of dSarmΔdsarm, which based on previous reports should rapidly degrade axonal NAD+ and normally leads to rapid axonal degeneration (Essuman, 2017), can be completely suppressed by loss of Axed. If the terminal step in axonal death were Nmnat2/Sarm1-dependent depletion of NAD+ pools, it is hard to imagine how the NAD+ hydrolase activity of the Sarm1 TIR domain, once unleashed, could be suppressed by the loss of a BTB and BACK domain protein (i.e., Axed). The alternative hypothesis is favored that Sarm1 signaling, perhaps through its TIR domain NAD+ hydrolase activity, activates an Axed-dependent downstream signaling pathway essential for axon death. ADP-ribosylation of targets is one mechanism that allows certain E3 ubiquitin ligases to bind substrates. There is certainly a tight association between NAD+ metabolism and axon death signaling, and the NAD+ hydrolase activity associated with the Sarm1 TIR domain can lead to the production of ADPr. Perhaps BTB and BACK domain proteins like Axed also require ADP-ribosylation of selected targets during axon death. In this model, the degradation of NAD+ would be a byproduct of the reaction, not a driving force (Neukomm, 2017).
Nmnat2-/- mutant mice die perinatally with neurons containing short axons, but these embryos can be rescued to adulthood by Sarm1-/- null mutations. It is likely that Nmnat2-/- animals and axons can survive in Sarm1-/- backgrounds because Nmnat1 and Nmnat3, the additional isoforms of Nmnat found in mice, could partially compensate. An nmnatRNAi experiment may therefore more closely resemble conditions in the Nmnat2-/-, Sarm1-/- mouse, with respect to the partial, but not complete elimination of Nmnat function. dsarm null alleles could only partially protect axons from Nmnat depletion with nmnatRNAi, as far fewer axons and cell bodies were observed compared to controls. Consistent with a requirement for Nmnat activity in cell survival, this study found null clones of the sole fly Nmnat molecule die within 1 day after eclosion. Remarkably, this phenotype can be completely overcome by loss of Axed, since axed, nmnat double mutant clones survive for weeks. However, it was surprising to find that loss of dSarm fails to block this degeneration. Whether Sarm1-/- animals could suppress the degeneration of mammalian neurons completely lacking Nmnat function would require the simultaneous elimination of all Nmnat isoforms (i.e., Nmnat1,2,3 triple mutants), which has not been explored. In the absence of Nmnat activity, invertebrate cells should not be able to autonomously synthesize NAD+. How these cells survive remains a mystery, but a number of possibilities might explain this phenotype. Perhaps yet-to-be-identified NAD+ biosynthetic pathways exist that can function without Nmnat. Alternatively, axons in vivo could be supplemented with NAD+ or key intermediates by surrounding glia and thereby sustain their integrity in axed, nmnat double mutant axons. Finally, it is possible that in the absence of Axed, Nmnat protein may not be turned over appropriately in axons and that perdurance of small amounts of Nmnat may generate sufficient levels of NAD+ for axon survival. Direct, in vivo measurement of NAD+ levels in axons in Drosophila will be essential to answer this question, but current NAD+ sensors have not yet provided sufficient sensitivity for this analysis (Neukomm, 2017).
Axed appears to sit genetically at a convergence point in axon death signaling. There are three ways to activate axon death: axotomy, Nmnat/Nmnat2 depletion, or expression of gain-of-function dSarm/Sarm1. Axed mutants, but not dsarm, can suppress each of these treatments, and, amazingly, axed mutants can even survive the combination of all three insults at once. The neuroprotective effects of axed therefore exceed those of dsarm mutants. It remains to be determined whether any of the four putative mammalian paralogs (BTBD1, BTBD2, BTBD3, and BTBD6) play a role in axon death, but this seems likely based on the strong conservation of dSarm/Sarm1 function in axon degeneration. If so, these would represent important new therapeutic targets for blocking axon death in neurological diseases such as traumatic brain injury, peripheral neuropathy, or nerve injury, where dSarm/Sarm1 signaling is known to drive axon loss (Neukomm, 2017).
Nervous system injury and disease have broad effects on the functional connectivity of the nervous system, but how injury signals are spread across neural circuits remains unclear. This study explored how axotomy changes the physiology of severed axons and adjacent uninjured "bystander" neurons in a simple in vivo nerve preparation. Within hours after injury, suppression of axon transport was observed in all axons, whether injured or not, and decreased mechano- and chemosensory signal transduction was observed in uninjured bystander neurons. Unexpectedly, it was found the axon death molecule Sterile alpha and Armadillo motif (dSarm), but not its NAD(+) hydrolase activity, was required cell autonomously for these early changes in neuronal cell biology in bystander neurons, as were the voltage-gated calcium channel Cacophony (Cac) and the mitogen-activated protein kinase (MAPK) signaling cascade. Bystander neurons functionally recovered at later time points, while severed axons degenerated via α/Armadillo/Toll-interleukin receptor homology domain (dSarm)/Axundead signaling, and independently of Cac/MAPK. Interestingly, suppression of bystander neuron function required Draper/MEGF10 signaling in glia, indicating glial cells spread injury signals and actively suppress bystander neuron function. This work identifies a new role for dSarm and glia in suppression of bystander neuron function after injury and defines two genetically and temporally separable phases of dSarm signaling in the injured nervous system (Hsu, 2020).
Nervous system injury or neurodegenerative disease can lead to profound alterations in neural circuit function. The precise cellular basis is poorly defined in any context, but disruption of circuit signaling is generally thought to occur as a result of a loss of physical connectivity between damaged neurons. Indeed, axon and synapse degeneration are among the best correlates of functional loss in patients with a variety of brain injuries or neurological diseases. But whether, and the extent to which, an injured or diseased neuron might also alter the functional properties of neighboring healthy 'bystander' neurons (i.e., those not damaged or expressing disease-associated molecules) is an important and open question. If the physiology of bystander neurons is radically altered by their damaged neighbors, this would force reconsideration of the simple loss-of-physical-connectivity model as the appropriate explanation for functional loss in neural circuits after trauma (Hsu, 2020).
It is well documented that bystander neurons can change their physiology in response to their neighbors being injured. For instance, mouse L5 spinal nerve transection results in the degeneration of distal L5 afferents in sciatic nerve alongside intact L4 C fiber afferents. Within 1 day after L5 lesion, L4 C fibers develop spontaneous activity that lasts for at least 1 week and appears to mediate injury-induced pain and hyperalgesia behaviors. Bystander effects have also been observed in the central nervous system (CNS). In a mouse model of mild traumatic brain injury (TBI), 1 day after injury, pyramidal neurons with severed axons and intact bystander neurons both exhibited injury-induced changes in action potential firing and afterhyperpolarization. Injured neurons failed to recover, while bystander neurons ultimately exhibited a return to normal firing properties. How injured neurons or surrounding glia signal to bystander neurons, or how bystander neurons receive this signal, is not known, but the similar electrophysiological changes observed in axotomized and intact dorsal root ganglion neurons have been proposed to be associated with Wallerian degeneration (Hsu, 2020).
Recent work has begun to illuminate the mechanisms by which damaged axons autonomously drive their own degeneration during Wallerian degeneration. A forward genetic screen in Drosophila identified the sterile α/Armadillo/Toll-interleukin receptor homology domain (dSarm) molecule as essential for axon auto-destruction, as loss of dSarm completely blocked Wallerian degeneration (Osterloh, 2012). All known dSarm pro-degenerative function requires the BTB and BACK domain molecule Axundead (Axed), another powerful regulator of axon degeneration (Neukomm, 2017). dSarm function in axon degeneration after injury is conserved in mouse: Sarm1-/- mutants block Wallerian degeneration, and loss of Sarm1 also suppresses axon degeneration in mouse models of TBI and peripheral neuropathy. Sarm1 inhibition is thus an exciting potential approach for blocking axon loss and neuroinflammation in human disease (Hsu, 2020).
dSarm/Sarm1 has been studied primarily in the nervous system as a positive regulator of axonal degeneration. In mammals, axotomy leads to the depletion of the labile NAD+ biosynthetic enzyme Nmnat2 and a decrease in NAD+ in severed axons. Nmnat2 loss somehow activates Sarm1 (Gilley, 2015), which is proposed to lead to further NAD+ depletion and metabolic catastrophe in the severed axon (Gerdts, 2015) through a Sarm1-intrinsic NAD+ hydrolase activity (Essuman, 2017). The Sarm1 NAD+ hydrolase activity appears to be activated directly by the NAD+ precursor, NMN, presumably through allosteric conformational changes in Sarm1 upon NMN binding. This NAD+ depletion model has been proposed as the primary mechanism by which Sarm1 drives axon loss, and to explain the mechanistic basis of protection by several other neuroprotective molecules (Gerdts, 2016). For instance, the slow Wallerian degeneration molecule (WldS), which includes the highly stable NAD+ biosynthetic enzyme Nmnat1, is thought to protect axons by substituting for the labile Nmnat2 molecule, thereby reducing NMN levels and avoiding NAD+ depletion. Similarly, the protective effects of loss of the E3 ubiquitin ligase Highwire/Phr1 is thought to result from blockade of its direct role in degrading Nmnat2, such that in hiw/phr1 mutants Nmnat2 is stabilized and continues to maintain NAD+ levels (Hsu, 2020).
Elegant genetic studies in C. elegans demonstrated that TIR-1 (the worm homolog of dSarm/Sarm1) is part of a signaling cascade downstream of the voltage-gated calcium channel UNC-36 and CamK-II and signals via the mitogen-activated protein kinase (MAPK) signaling cascade. Based on this work, MAPK signaling was examined for roles in Wallerian degeneration but met with mixed results. Changes in MAPK signaling (i.e., phosphorylation of MAPK pathway members) were found in axons within 15-30 min after axotomy, were Sarm1 dependent and suppressed by Nmnat overexpression, and partial suppression of axon degeneration was observed after simultaneous blockade of multiple MAPK components. But how MAPK signaling modulates axon degeneration, particularly in the context of Sarm1 signaling, remains controversial, as one study proposed MAPK signals downstream of Sarm1, while another argued Sarm1 was upstream of MAPK signaling, and the neuroprotective phenotypes resulting from MAPK blockade do not approach levels afforded by loss of Sarm1 in vivo (Hsu, 2020).
This study used a partial nerve injury model to examine early changes in the physiology of severed axons and neighboring uninjured bystander neurons. Axotomy of even a small subset of neurons was shown to leads to inhibition of cargo transport in all axons within the nerve and suppression of sensory signal transduction in bystander neurons. Surprisingly, this early blockade of axon transport and sensory signal transduction required dSarm in both severed and uninjured bystander neurons, where it signaled via the conserved UNC-36/MAPK signaling pathway. Early suppression of axon transport and bystander neuron function did not require dSarm NAD+ hydrolase function or Axed, was not modulated by NMN, and was not induced by depletion of dNmnat. This suggests it is mechanistically different from later events in axon death, where dSarm drives axon degeneration with Axed. Intriguingly, this study found that this early spreading of injury signals to bystander neurons required the Draper receptor in surrounding glia, indicating that glial cells actively signal to inhibit the function of bystander neurons in vivo. This work identifies new roles for dSarm and glia in modifying neurophysiology early after injury, assigns the NAD+ hydrolase function exclusively to later axon degenerative events, and reveals a new role for UNC-36/MAPK signaling in promoting these dSarm-dependent changes early after an injury has occurred in the nervous system. It is proposed that two temporally and genetically separable phases of dSarm signaling exist that mediate these distinct injury-induced changes in neurophysiology and axon degeneration (Hsu, 2020).
This study shows that relatively small injuries can lead to the rapid and efficient spreading of injury signals across nerves that potently suppress axon transport throughout the nerve, and broadly inhibit neurophysiology in uninjured bystander neurons. Surprisingly, the same molecule was found to be required to drive explosive axon degeneration in severed axons at later stages, dSarm/Sarm1, is required for this early suppression of neuronal function, although the signaling mechanisms at each stage appear to be different. The data support a model whereby early (i.e., 1-3 h after injury) dSarm signals with Cac and MAPK components, but independent of its NAD+ hydrolase activity, to suppress axon transport and neurophysiology, while at later stages (8-12 h), dSarm signals with Axed to promote explosive axon degeneration. This significantly expands the role for dSarm/Sarm1 in regulating nervous system responses to injury to include even uninjured bystander neurons. Furthermore, a critical role was discoverd for glial cells, through Draper, in signaling to bystander neurons to inhibit their axon transport and neurophysiology. Together, this work suggests that a significant amount of functional loss after neural trauma is a result of not only frank degeneration but also more widespread changes in neuronal function, and it occurs in uninjured neurons through glial spreading of injury signals (Hsu, 2020).
The data support the notion that widespread signaling occurs between cells in injured neural tissues immediately after injury and that injury signals can radically alter neuronal function. Severing even a small number of axons led to a suppression of axon transport within hours in all axons in the adult wing nerve, even in uninjured bystander neurons. Beyond axon transport, local uninjured bystander sensory neurons also exhibited a disruption of mechano- and chemosensory signal transduction, which was partially reversible within a few hours. These observations suggest that beyond simple breakage of connectivity, a significant part of functional loss after brain injury or in neurodegenerative disease may also be occurring in healthy, intact neurons that have received function-suppressing signals from nearby damaged neurons (Hsu, 2020).
Surprisingly, dSarm was found to be required cell autonomously in bystander neurons to alter axon transport and nerve function in response to injury, and this role did not require its NAD+ hydrolase activity. Reception of this injury signal in the bystander neuron (and severed axons) requires the VGCC Cac and the MAPK signaling cascade, similar to Tir-1 signaling in C. elegans, but not Axed. Reciprocally, Cac and MAPK components are not required for Wallerian degeneration at later stages. Explosive axon degeneration requires dSarm, its NAD+ hydrolase activity, and Axed. Based on the timing of these different events (i.e., changes in neuronal function versus frank degeneration) with the genetic studies indicating they are separable, a two-phase model is proposed for dSarm signaling in injured neural tissues: early dSarm-dependent changes in axon biology and neurophysiology that occur within hours after injury are mediated by the Cac/dSarm/MAPK signaling cascade (phase I), while late-stage axon degeneration is driven by dSarm signaling through Axed (phase II). The existence of these temporally distinct phases of dSarm signaling likely explain previous results that seemed in conflict, where MAPK signaling was proposed to act both upstream (Yang, 2015) and downstream (Walker, 2017) of Sarm1 after axotomy. According to the current model, both of these assertions would be correct, with dSarm/Sarm1 acting upstream of MAPK early (phase I) and independent but ultimately downstream of MAPK later to drive dSarm/Axed-dependent axon degeneration (phase II)(Hsu, 2020).
To date, dSarm/Sarm1 has been thought of primarily as a cell-autonomous regulator of explosive axon degeneration, but the current work shows that dSarm can also drive important changes in circuit function through altering neuronal cell biology and neurophysiology. That bystander neurons recover and remain viable also demonstrates that activation of dSarm after injury does not necessarily lead to axon death. It is suspected that recovery occurs in large part because bystander neurons have not been severed, which is an extreme injury, and depends on their connection to the cell body, which is a source of axon survival factors like Nmnat2. Connection to the cell body may also explain why axon transport was less severely suppressed in the bystander neurons; additional transport factors can still be continuously supplied to the distal axon from the soma. Defining how dSarm activity is regulated in each of these contexts to interact with Cac/MAPKs versus Axed, and why the first phase does not require NAD+ hydrolase function, are key questions for the future (Hsu, 2020).
A compelling case exists for the NAD+ depletion hypothesis for dSarm/Sarm1 function in axon degeneration (Essuman, 2017; Gerdts, 2015, 2016), although arguments have been made this dSarm/Sarm1 signaling is likely more complex (Neukomm, 2017). In this model, depletion of Nmnat2 via Hiw/Phr1 results in the accumulation of NMN, which functions as an activator of Sarm1, with Sarm1 NAD+ hydrolase activity driving metabolic catastrophe. This study provides several lines of evidence that the above, newly described early dSarm signaling events (i.e., suppression of axon transport and neurophysiology) are mechanistically distinct but are nevertheless also regulated by some axon-death-associated molecules. First, while NMNd can suppress axon degeneration in flies and other species, it cannot block early suppression of axon transport or changes in bystander neuron function. This argues that NMN is not a driving force for dSarm activation in the early phase. Second, although limited to tagged versions of dNmnat for this analysis, no depletion of dNmnat was observed within the time frame of 6 h after injury. Previous studies in SCG or DRG cultures in vitro suggest Nmnat2 depletion takes 4-6 h and NAD+ depletion begins ~2-3 h after axotomy, which is slightly later than the bystander effect was observed in vivo. Because full axon degeneration is prolonged in vivo compared to in vitro studies, the timing of Nmnat2 loss and NAD+ depletion is likely also prolonged in vivo, further suggesting this likely happens after cessation of axon transport. Third, Axed, which is genetically downstream of dSarm during axon degeneration (Neukomm, 2017), is not required for early suppression of nerve responses to injury in either severed or intact neurons, only later axon degenerative events in the severed axons. Finally, this study shows that while the NAD+ hydrolase function of dSarm is required in vivo for efficient axon degeneration, it is dispensable for early suppression of axon transport (Hsu, 2020).
Despite these clear molecular and genetic differences between early- and late-phase signaling events, WldS or dNmant expression or hiw mutants are capable of suppressing early changes in axon transport and neurophysiology, even in bystander neurons. This could be interpreted as evidence for similarity in signaling mechanisms at early and late stages of dSarm signaling (i.e., that they act by maintaining NAD+). However, the alternative possibility is favored that these data point to an important role for dNmnat in mediating early dSarm signaling events during suppression of bystander neuron function. Loss of Axed does not affect the bystander effect, and axon transport is suppressed. However, this study found that loss of dNmnat in axed null backgrounds (which allows for preservation of neuronal integrity despite loss of dNmnat) blocked the ability of injury to induce the bystander effect. This result reveals a paradoxical, positive role for dNmnat in promoting the bystander effect early. It is suspected that dNmnat exerts this effect through modulating MAPK signaling, whose interactions are complex: loss of Nmant has been shown to suppress MAPK signaling, while increased Nmnat activity can also potently block the activation of MAPK signaling within the first few hours after axotomy. It is proposed that dNmnat activity is required early for the bystander effect and that dNmnat levels need to be precisely tuned for proper signaling at each phase (Hsu, 2020).
Glial cells are well positioned to rapidly spread signals to all axons in the wing nerve. Much like Remak bundles in mammals, the Drosophila L1 wing nerve has glial cells that appear to wrap axons individually, which would imply that axon-to-axon signals must pass through glia. The observation that selective elimination of Draper signaling in glia is sufficient to inhibit the spreading of injury signals to bystander neurons is consistent with an axon->glia->bystander neuron signaling event, although it is also possible that glia are directly injured by the axotomy and signal to bystander neurons without input from the severed axons. Given the similarities in the response of severed axons and those of bystanders (i.e., both block axon transport on the same timescale), and the selective effects of Draper on the bystander neuron axons, the former model is favored rather than the latter (Hsu, 2020).
Draper signals to bystander neurons through a transcriptional JNK/dAP-1 cascade, likely through activating MMP-1. Nerve injury also rapidly activates JNK/c-Jun signaling in mammalian Schwann cells, where JNK/c-Jun mediate most aspects of Schwann cell injury responses and reprogramming events. This conserved glial response likely occurs in Schwann-cell-like wrapping glia present in the Drosophila L1 wing nerve, although it may be activated in the subperineurial glia, which can act in a partially redundant fashion with wrapping glia. The involvement of Mmp-1 is intriguing given its well-known role in neuroinflammatory responses to brain injury in mammals, where it functions to break down the extracellular matrix and has been proposed to promote diffuse axon injury. Other key components of the Draper signaling pathway (dCed-6 in particular, which is required for Draper signaling in all other known contexts) were not required for suppression of bystander neuron neurophysiology (Hsu, 2020).
How bystander neurons receive injury signals and respond has remained unclear, although injury- or disease-induced effects on bystander neurons is well documented. In most cases, this has been explored in the context of bystander neuron cell death driven by neuroinflammatory cells. For instance, release of C1q, interleukin-1α (IL-1α), tumor necrosis factor (TNF) from microglia following brain injury drives the formation of neurotoxic astrocytes, which can promote the death of neurons through release of yet-to-be-identified toxins. Bystander neuronal cell death is also driven by brain-infiltrating inflammatory monocytes in viral encephalitis, in a way that is mediated by calpains, which are also important regulators of axon degeneration. Secondary axon degeneration (i.e., that occurring in neurons not damaged by the initial injury) can be driven in a way that requires intracellular Ca2+ release through IP3Rs and ryanodine receptors. These represent extreme cases of bystander effects, where cells undergo apoptosis or their axons degeneration. Whether dSarm/Sarm1 is involved in these effects is an open question. The model employed by this study is likely most relevant to partial nerve injury, where non-autonomous changes in bystander neurons have been well documented. Uninjured bystander neurons in mild TBI models are certainly altered physiologically in a reversible way. The molecular basis of any of these signaling events remains unknown, but this study points to dSarm/Sarm1 as a candidate mediator. It is interesting to note that in contrast to control mice, which show significant behavioral defects for hours after mild TBI, Sarm1-/- animals exhibited almost immediate recovery, and this was at a time point long before diffuse axon injury is observed in TBI models. It is plausible that this early loss of function is mediated in part by the bystander effect (Hsu, 2020).
In summary, this study defines two genetically separable phases of dSarm signaling, places dSarm/Sarm1 at the heart of neuronal injury signaling throughout neural tissues, identifies new signaling partners for dSarm, and expands its role to regulating the responses of uninjured neurons to local tissue injury (Hsu, 2020).
Neurodegenerative stimuli are often associated with perturbation of the axon initial segment (AIS), but it remains unclear whether AIS disruption is causative for neurodegeneration or is a downstream step in disease progression. This study demonstrates that either of two separate, genetically parallel pathways that disrupt the AIS induce axonal degeneration and loss of neurons in the central brain of Drosophila. Expression of a portion of the C-terminal tail of the Ank2-L isoform of Ankyrin severely shortens the AIS in Drosophila mushroom body (MB) neurons, and this shortening occurs through a mechanism that is genetically separate from the previously described Cdk5alpha-dependent pathway of AIS regulation. Further, either manipulation triggers morphological degeneration of MB axons and is accompanied by neuron loss. Taken together, these results are consistent with the hypothesis that disruption of the AIS is causally related to degeneration of fly central brain neurons, and it is suggested that similar mechanisms may contribute to neurodegeneration in mammals (Spurrier, 2019).
The axon initial segment (AIS) is a key neuronal domain that lies between the somatodendritic and axonal compartments. The AIS is a gatekeeper for intracellular transport that maintains neuronal polarity, and it is essential for proper neuronal excitability as it is the site of action potential (AP) initiation and a critical target for AP modulation. The AIS performs these tasks by virtue of a unique composition of membrane proteins, including a distinctive set of voltage-gated ion channels and submembranous cytoskeletal proteins. The structure of the AIS in vertebrate neurons depends on the presence of a 'master organizer,' Ankyrin G (ankG), a giant ankyrin isoform that recruits and anchors the specialized set of proteins that performs its distinctive functions (Spurrier, 2019).
Aberrant AIS regulation and function have been linked to a variety of diseases. Changes in sequence or expression levels of proteins localized to the AIS are involved in epilepsy, bipolar disorder, schizophrenia [, and autism spectrum disorder. Perturbation of the AIS has also been linked to neurodegeneration under a variety of contexts. The AIS contributes to proper tau localization, and breakdown of its barrier function leads to tau missorting, while expression of Alzheimer's disease (AD)-related tau mutants was found to decrease AIS-associated proteins and shorten the overall length of the AIS, SPTBN4 (encoding non-erythrocyte β-spectrin 4) was identified in a DNA methylation screen of genes related to AD; β-spectrin works in coordination with ankG to localize proteins to the AIS. Further, in a mouse model of AD, it was found that AIS length was reduced near Aβ plaques. Aβ plaques are also associated with microglia recruitment, which is itself associated with AIS structural plasticity. While recent literature suggests that various provocations, including pathology, can trigger structural plasticity of the AIS, the underlying mechanisms remain enigmatic (Spurrier, 2019).
Previous work has shown that some Drosophila neurons include a domain that exhibits the characteristic hallmarks of the mammalian AIS, providing a simpler model in which to study the molecular pathways contributing to AIS biogenesis and regulation (Trunova, 2011). First, there is selective accumulation of an anchoring protein within this subcellular domain. Drosophila only has two ankyrin genes: Ank1, which is expressed ubiquitously, and the neuron-specific Ank2. While Ank1 is expressed in all cells, it is present at elevated levels in the AIS of MB neurons (Trunova, 2011). Second, the Drosophila AIS contains a unique combination of voltage-gated potassium channels. Specifically, Elk, Shaw (Kv3), and Shal (Kv4) channels were enriched in the AIS, while dORK-C2 (Ork1), Shaker (Kv1.3), and EKO (Kv1.3) were selectively excluded. Lastly, there is an altered F-actin cytoskeleton within the AIS. In the somatodendritic and axonal regions, actin is highly expressed and ubiquitous; within the AIS, actin levels are reduced significantly and appear to have a distinctively patterned distribution. An AIS-like domain has also been observed in multipolar dendritic arborization neurons of flies. Jegla (2016) identified a diffusion barrier localized to the proximal axon of ddaE neurons that coincided with the enrichment of Shal and Elk potassium channels. Furthermore, they found that a fragment of Ank2-L was targeted to the proximal axon in the same area. As Ank2-L is one of the two Ank2 isoforms that have giant exons that share structural similarities with AnkG, this suggests that Ank2-L is an analog, and possibly an ortholog, of mammalian AnkG (Spurrier, 2019).
Similar to the mammalian AIS, the AIS in flies is also linked to neurodegenerative stimuli. Deletion of Cdk5α (also called D-p35), encoding the activating subunit for cyclin dependent kinase 5 (Cdk5), has been shown to induce multiple degenerative phenotypes in Drosophila central brain neurons, including impaired autophagy, swelling of proximal axons, histologically-evident tissue loss (i.e., formation of 'vacuoles' in the brain), and age-dependent loss of neurons. Cdk5α-null flies also exhibit an AIS that is severely shortened and indeed nearly absent: though a barrier remains that separates the somatodendritic and axonal compartments, the domain is so short that the characteristic pattern of accumulation of molecular markers cannot be detected. Notably, the position of the missing AIS correlates with the location where axonal swellings form in affected neurons and where histological tissue loss is observed. However, it remains unclear if disruption of the AIS merely correlates with neurodegeneration or plays a causative role (Spurrier, 2019).
This study shows that overexpression of a portion of the carboxyl domain of the Ank2-L isoform results in a severely shortened AIS in Drosophila central brain neurons, reminiscent of that observed in Cdk5a-null. Ank2-L4-mediated modulation of the AIS, however, occurs through a pathway that is genetically parallel to the previously identified Cdk5α-dependent mechanism. Strikingly, dysregulation of either pathway results in morphological degeneration of axons, and is associated with neuron loss. Therefore, an orthogonal molecular manipulation that shortens the AIS, acting by a genetically independent pathway, also causes axonal degeneration, just as was observed with altered Cdk5α. This provides evidence supporting the hypothesis that disruption of the AIS is apt to be causal for degeneration in fly MB neurons, and may contribute to neurodegeneration in mammalian disease (Spurrier, 2019).
Previous experiments have shown that defects in the axon initial segment are sometimes observed in neurons that have been subjected to neurodegenerative insults, but whether the AIS defects are themselves causal for degeneration has remained unknown. This study has identified a novel reagent to manipulate the AIS in Drosophila central brain neurons. Overexpression of a C-terminal portion of the Ank2-L isoform drastically shortens the AIS in MB neurons, as measured with multiple markers, and that this modulation of the AIS occurs through a genetic pathway that is parallel to Cdk5α-mediated regulation of the AIS. Further, this study demonstrated that dysregulation of either mechanism triggers axonal degeneration, and in some cases, age-dependent loss of MB neurons (Spurrier, 2019).
Based on structural similarities and the presence of a giant exon, Drosophila Ank2-L has been proposed to be the ortholog of the mammalian AnkG and serves as a master organizer of the AIS. The current results confirm an important role of Ank2-L for AIS formation and maintenance but suggest that other components can compensate for its function in regulating the size of the AIS, at least in part. In mammals, deletion of AnkG results in the complete loss of the AIS. Consistent with this, a partial shortening of the AIS was observed when Ank2-L levels were reduced, either genetically, with a null mutant, or with RNAi. However,
complete absence of the AIS from reduced Ank2-L levels was not seen, even when those levels are decreased enough to cause lethality. This discrepancy could stem from the differing sets of ankyrin genes between flies and mammals. Drosophila only has two identified ankyrins while mammals carry three ankyrin genes. It may be that as mammals have an expanded ankyrin repertoire with more specialized functions for each gene, the loss of one completely ablates the associated function. In Drosophila, the neuron-specific Ank2 is accompanied by Ank1, which is also enriched at the AIS. It is possible that deletion of Ank2 disrupts the AIS to a certain degree, but there is compensation by Ank1, or by another Ank2 isoform, such as Ank2-XL, preventing the complete dissolution of the AIS. Interplay between multiple ankyrins at the AIS has been shown in mammals, as overexpression of AnkB in cultured hippocampal neurons resulted in a more restricted distribution of AnkG at the AIS. Another possibility is that Ank1 plays a significant role in the organization of the Drosophila AIS. However, Ank1 lacks the giant exons common to mammalian AnkG, so if it were to be capable of acting as the sole organizer of the AIS, it would likely do so through somewhat different mechanisms. Experiments reducing Ank1 levels selectively in the MB will be informative to reveal the specific contributions of Ank1 in establishment and maintenance of the AIS. Furthermore, it will be interesting to see if Ank1 is altered in MB neurons when Ank2-L levels are perturbed or Ank2-L4 is overexpressed (Spurrier, 2019).
It was shown recently that Ank2-L localizes to the AIS in multipolar ddaE neurons, and Ank2-null mutants exhibit loss of a diffusion barrier and loss of potassium channel localization in these neurons (Jegla, 2016). One potential explanation for the apparent difference in AIS phenotype in MB vs ddaE neurons upon Ank2-deletion could be cell type differences. At the neuromuscular junction (NMJ) of Drosophila motoneurons, Ank2-L was localized to the axon and pre-synaptic terminal. In ddaE neurons, in contrast, Ank2-L4 accumulated in the proximal axon and regulated the diffusion barrier, while Ank2-S appeared ubiquitously throughout the entire neuron. In the current study, neither Ank2-S, Ank2-L4, nor Ank2-L8 showed any specific localization within MB gamma neurons, as accumulation of all three was observed in the cell bodies, dendrites, and axonal lobes. Thus, it seems that different neuronal subtypes may use different targeting paradigms. A second possible explanation for the differences between these results could be the sensitivity of the assays used to measure AIS defects. Measuring changes in the boundary of antibody staining is largely qualitative, whereas intermediate effects might be easier to recognize when assaying a quantitative measure, such as diffusion rates. Regardless, both the current dzta and the study by Jegla (2016). support the conclusion that Ank2 contributes to the proper maintenance of the axon initial segment (Spurrier, 2019).
Overexpression of the C-terminal portion of Ank2-L drastically shortened the AIS, as did genetic reduction or removal of Ank2-L. This was initially unexpected, as overexpression of AnkG in cultured hippocampal neurons nearly tripled the length of the AIS, and suggests that the isolated C-terminal domain of Ank2-L acts as a dominant negative and not as a gain of function. Indeed, expression of the Ank2-L C-terminus had a significantly stronger effect on the AIS than even the Ank2-L null mutant, a property known genetically as an 'antimorph'. It may be, for example, that Ank2-L4 acts by mislocalizing the normal binding partners of Ank2-L, either directly or by changing the localization of the endogenous Ank2-L, or that it mislocalizes or inactivates components that would normally be able to compensate for the absence of Ank2. Formally, it cannot be rule out that Ank2-L4 acts as a neomorph with regard to AIS regulation, as the total absence of Ank2-L in homozygous mutants failed to cause the severely shortened phenotype seen with Ank2-L4 overexpression. It has been demonstrated previously that the complete C-terminal domain of Ank2 exhibits neomorphic activity during neuromuscular junction development, as expression of Ank2-L8 in presynaptic neurons at the NMJ results in the formation of small, highly ramified satellite boutons at the synapse. It is also striking that the antimorphic effect of Ank2-L is assay dependent; the AIS phenotype is stronger than that of the null mutant, but unlike the genetic mutant, the Ank2-L C-terminus does not induce lethality even when expressed pan-neuronally at high level. Such assay-specific differences in phenotypic strength are not uncommon, and may arise, for example, from differences in the spectrum of proteins that bind a particular domain in different tissues (Spurrier, 2019).
The cell loss observed from expression of Ank2-L was far less extensive than the axonal morphological phenotypes. This finding raises questions about the relationship of axonal degeneration to loss of the cell soma. Deletion of Cdk5α results in a shortened AIS, as well as causing degenerative phenotypes and neuronal loss that stem, in part, from an acceleration of physiological aging and hyperactivation of the immune system. However, the level of neuron loss in Cdk5α-null flies was greater than what was observed in this study. This suggests that some aging-associated degenerative pathways are more strongly productive of soma loss, while axonal degeneration, which one would expect to be equally effective at disrupting neuronal function, may not always lead to death of the soma, at least in Drosophila (Spurrier, 2019).
Disruption of the AIS has been observed in association with multiple neurodegenerative stimuli, including altered expression of Cdk5α in Drosophila and tau mutation or acetylation in mic]. The data in this study show that a manipulation that directly targets a structural component of the AIS leads to axonal degeneration, as Ank2-L4 overexpression resulted in a significant increase in the prevalence of axonal swelling within the proximal axon, and blebbing of the axon in aged flies, in concert with shortening the AIS. Additionally, Ank2-L4 overexpression resulted in a significant increase in cell loss relative to control. The observed degenerative phenotypes and cell loss do not appear to be due to non-specific detrimental effects on neuronal morphology or physiology during development. Expression of neither Ank2-L4 nor Ank2-L8, which had a more severe effect on the AIS, significantly altered the gross morphological structure of the MB neurons of 3rd instar larva, indicating that any phenotypes are largely restricted to subcellular organization. Moreover, it is worth noting that even though the AIS was shortened to the point that it could not be detected with molecular markers when Ank2-L4 was overexpressed, Futsch remained restricted to the somatodendritic region suggesting maintenance of a diffusion barrier, just as was observed in Cdk5α-null mutant flies. Thus, it is unlikely that the observed neurodegeneration stems from gross disruption of neuronal polarity or morphology (Spurrier, 2019).
The data reported in this study show that the structure of the AIS is regulated by at least two parallel pathways, one defined by Cdk5α and one by Ank2-L, and that perturbation of either pathway leads to axonal degeneration and neuron loss. As such, the most parsimonious interpretation is that a shared feature, such as the altered AIS, is responsible for the observed degenerative phenotypes. However, it cannot be excluded that overexpression of the dominant-negative Ank2-L4 disrupts some other aspect(s) of cell structure or physiology that it also shares with Cdk5α. Modest structural plasticity of the AIS does not always lead to degeneration; indeed, modulation of AIS length is a vital part of fine-tuning neuronal excitability. Thus, shortening of the AIS by overexpression of Ank2-L4 may be only one aspect of disturbed AIS function and neuronal physiology from this manipulation. For example, the data also demonstrate alteration of actin organization in the AIS, and the localized swelling of the axon is likely associated with defects in axonal transport. Moreover, the drastic reduction observed in the extent of the AIS is expected to have severe consequences for neuronal excitability. Finally, Ank2-L4 expression may cause unrecognized changes in other parts of the cell. The key point, however, is that the data seems to exclude the hypothesis that AIS loss is simply a secondary, late-stage marker of neurodegeneration, but rather suggests that it is associated with the earliest stages of the process, as targeting either of two independent regulators of the AIS, one a core structural component, is sufficient to induce degeneration (Spurrier, 2019).
Using a novel reagent, this study has unlocked a new method for modulating the AIS independent of Cdk5/Cdk5α, the only known AIS regulator in Drosophila MB neurons. The regulation of the AIS presented in this study is likely to be direct as Ank2-L4 acts as an antimorph of the essential ankyrin isoform that helps construct the AIS. Remarkably, this study showa that a genetic treatment that disrupts the AIS by manipulation of one of its core components is also sufficient to cause axon degeneration and even neuron loss in flies, supporting the hypothesis that the AIS disruption observed in association with several neurodegenerative mechanisms contributes causally to the process of neurodegeneration (Spurrier, 2019).
The structural integrity of synaptic connections critically depends on the interaction between synaptic cell adhesion molecules (CAMs) and the underlying actin and microtubule cytoskeleton. This interaction is mediated by giant Ankyrins, that act as specialized adaptors to establish and maintain axonal and synaptic compartments. In Drosophila, two giant isoforms of Ankyrin2 (Ank2) control synapse stability and organization at the larval neuromuscular junction (NMJ). Both Ank2-L and Ank2-XL are highly abundant in motoneuron axons and within the presynaptic terminal, where they control synaptic CAMs distribution and organization of microtubules. This study addresses the role of the conserved N-terminal ankyrin repeat domain (ARD) for subcellular localization and function of these giant Ankyrins in vivo. A P[acman] based rescue approach was used to generate deletions of ARD subdomains, that contain putative binding sites of interacting transmembrane proteins. Specific subdomains control synaptic but not axonal localization of Ank2-L. These domains contain binding sites to L1-family member CAMs, and these regions are necessary for the organization of synaptic CAMs and for the control of synaptic stability. In contrast, presynaptic Ank2-XL localization only partially depends on the ARD but strictly requires the presynaptic presence of Ank2-L demonstrating a critical co-dependence of the two isoforms at the NMJ. Ank2-XL dependent control of microtubule organization correlates with presynaptic abundance of the protein and is thus only partially affected by ARD deletions. Together, these data provides novel insights into the synaptic targeting of giant Ankyrins with relevance for the control of synaptic plasticity and maintenance (Weber, 2019).
Giant ankyrins serve as central organizing adaptor molecules in both invertebrate and vertebrate neurons. This study provides first insights into the control of synaptic localization and function of giant Ankyrins by the N-terminal ankyrin repeat domain in vivo. By selectively deleting subdomains of the ARD of ank2 this study has unravel critical requirements of specific regions of the ARD for the synaptic localization of the neuronal specific giant isoforms Ank2-L and Ank2-XL. The data demonstrate that the N-terminal domain controls not only synaptic targeting of individual isoforms but also contributes to synaptic localization of the alternative isoform. The functional requirements of Ank2-L and Ank2-XL for synaptic stability and microtubule organization clearly correlate with ARD-dependent regulation of protein abundance at the presynaptic terminal, with individual subdomains providing unique functional features (Weber, 2019).
The N-terminal ARD mediates membrane-association of Ankyrins and is essential for the subcellular localization and organization of transmembrane binding partners. Prior work largely focused on the organizational roles of giant Ankyrins at the AIS and nodes of Ranvier in vertebrate neurons. The current in vivo analysis of ARD deletions now revealed a critical importance of this domain for the localization of Ank2-L at the presynaptic terminal of motoneurons. The synaptic localization of Ank2-L depends on AnkD2-4 (repeats 7-24) while the first domain (repeats 1-6) only had a minor impact on protein abundance. Interestingly, axonal targeting of Ank2-L was only partially affected by these deletions. These results indicate that separate and distinctive mechanisms exist in vivo that enable selective localization of giant Ankyrins in axons and within the presynaptic motoneuron terminal. As similar localization defects are observed already at earlier stages of development argues that AnkD2-4 are essential for initial synaptic localization. It was previously demonstrated that a fusion protein comprising the specific C-terminal tail domain of Ank2-L efficiently localizes to both axonal and synaptic areas (Stephan, 2015). The demonstration that synaptic localization of full length Ank2-L requires an intact ARD indicates that subcellular distribution is precisely regulated and not achieved by passive distribution within the neuron. Of particular interest is the observation that the first six ankyrin repeats are dispensable for synaptic localization of Ank2-L in the ank2ΔL mutant background. The recent characterization of the structure of the human ARD demonstrated distinct and independent binding sites for voltage-gated sodium channels (Nav1.2) and for L1-family CAMs (Neurofascin) in vitro and in cultured neurons. Interestingly, in cultured hippocampal neurons the L1-family CAM binding sites within AnkD2 (ankyrin repeats 8-9 and 11-13) are essential for clustering of the 270 kDa AnkG isoform at the AIS. In contrast, AnkG lacking the Nav1.2 binding site within AnkD1 (ankyrin repeats 2-6) efficiently localized to the AIS but failed to cluster Nav channels at the AIS. Thus, similar to the situation at the vertebrate AIS, the synaptic localization of Drosophila Ank2-L largely depends on interactions with L1-family CAMs or alternative transmembrane proteins that occupy binding sites within the central and C-terminal part of the ARD. This localized L1-CAM-Ankyrin complex can then serve as an assembly platform for additional molecules like voltage-gated sodium channels that acquired Ankyrin-binding capacities during chordate evolution to determine the physiological properties of specific subcellular neuronal compartments. While the AnkD1 of Drosophila ank2 had the least impact on synaptic stability phenotypes it will be interesting to identify putative binding proteins that may provide specific functional properties as absence of this domain resulted in decreased survival and locomotion of adult flies (Weber, 2019).
In contrast to Ank2-L, the deletion of individual parts of the ARD had smaller effects on synaptic targeting of Ank2-XL in the ank2ΔXL mutant background. Here, only the deletion of AnkD3 (repeats 13-18) resulted in a significant reduction of axonal and synaptic localization. However, this analysis revealed a striking dependence of Ank2-XL on the presence of the intact ARD of Ank2-L. Impairments of Ank2-L localization, including the minor effects caused by deletion of AnkD1, resulted in a severe reduction of synaptic Ank2-XL levels. This shows that Ank2-XL localizes via Ank2-L to the presynaptic terminal consistent with prior observations (Stephan, 2015). Interestingly, deletions of parts of the ARD of Ank2-XL also significantly reduced synaptic Ank2-L levels indicating a critical co-dependence of Ank2-L and Ank2-XL at the NMJ. This interaction may depend on direct interactions between the two Ankyrin isoforms, potentially mediated within the ARD. However, it is equally possible that incorporation of mutated Ankyrins with altered binding properties resulted in a disruption of the larger Ankyrin-Spectrin scaffold that in turn leads to reduced targeting of the other isoform. Indeed, in vertebrate axons it has been demonstrated that different affinities of specific giant Ankyrins control the isoform-specific incorporation into selective axonal compartments like the nodes of Ranvier (Weber, 2019).
At a functional level a strong correlation was observed between synaptic Ank2-L level and the control of synaptic stability. At all muscle groups analyzed deletion of AnkD1 domain had the mildest impact on synaptic stability consistent with the least impairments of synaptic Ank2-L localization. Importantly, the analysis of synaptic stability at muscle 4 also revealed that AnkD3 is most critical for synaptic maintenance. Despite an almost identical reduction in Ank2-L level compared to AnkD2 and 4 the rescue construct lacking the third domain was unable to restore any synaptic stability of ank2ΔL mutant animals. Interestingly, this assay also highlighted the specificity of the individual ankyrin repeats within the ARD. The exchange of second domain sequences with analogous sequences of the first domain failed to restore both Ank2-L localization and the synaptic stability phenotype of ank2ΔL mutants. The failure to support synaptic stability is at least in part due to a failure to organize and stabilize synaptic cell adhesion molecules. This analysis demonstrated that AnkD2 and 3 are critical for normal clustering of the NCAM homolog Fas II and of the L1 CAM homolog Nrg. This is consistent with a prior observations that Ank2-L supports synaptic organization of both Fas II and Nrg and the co-dependence of Ank2-L and these CAMs at the synapse (Pielage, 2008; Enneking, 2013). This function is evolutionary conserved as mutations in the second and third binding site of vertebrate AnkG failed to restore clustering of the L1 CAM family members Neurofascin or Nr-CAM at the AIS of hippocampal neurons (Weber, 2019).
In contrast to the clear requirements of the ARD for presynaptic targeting of Ank2-L the same mutations only partially affected localization of Ank2-XL. Consistently, only mild disruptions of the presynaptic microtubule cytoskeleton and of the synaptic bouton organization compared to the ank2ΔXL mutation were observed. The effects were most severe when deleting AnkD2 despite the fact that Ank2-XL protein levels were more compromised in AnkD3 mutants. As the large C-terminal repeat region of Ank2-XL is essential for the interaction with microtubules these results indicate that inappropriate ARD-dependent complex assembly may influence the functional properties of Ank2-XL at the NMJ (Weber, 2019).
Together, this in vivo analysis of the ARD of Drosophila giant Ankyrins uncovered a structural basis for presynaptic localization and provided a genetic basis for the identification of regulatory mechanisms controlling structural synaptic plasticity via the selective Ankyrin complex assembly (Weber, 2019).
Promoting axon regeneration in the central and peripheral nervous system is of clinical importance in neural injury and neurodegenerative diseases. Both pro- and anti-regeneration factors are being identified. Previous work has shown that the Rtca mediated RNA repair/splicing pathway restricts axon regeneration by inhibiting the nonconventional splicing of Xbp1 mRNA under cellular stress. However, the downstream effectors remain unknown. Through transcriptome profiling this study has shown that the tubulin polymerization-promoting protein (TPPP) ringmaker/ringer In recent years, several strategies have shown efficacy augmenting nerve regeneration in various experimental models. Unfortunately, therapeutic interventions to promote nerve regeneration and functional recovery still do not exist. Previous work has also helped shape the approach researchers have taken toward better understanding regeneration and drawing connections between successful paradigms. This study reports a link between two cellular mechanisms that are essential for regeneration: RNA processing and microtubule dynamics (Monahan Vargas, 2020).
In Drosophila, sensory dendritic arborization (da) neurons show differential regenerative potentials between the periphery and the central nervous system (CNS), resembling that of mammalian neurons. Moreover, distinct subclasses of da neurons also regenerate differently. A previous study developed a two-photon-based axon injury model that assays class III (C3da) and class IV (C4da) da neurons to identify and analyze targets that enhance regeneration. Using this model, Rtca (RNA 3'-terminal phosphate cyclase), an RNA-binding protein (RBP), was identified as an inhibitor of axon regeneration. Rtca is involved in stress induced Xbp1 mRNA splicing, and its knockout or neuronal knockdown promotes axon regeneration both in the peripheral nervous system (PNS) and CNS. However, its downstream effectors and signaling mechanisms remain unexplored. RBPs are increasingly shown to regulate complex cellular processes associated with neurodegenerative diseases and regeneration. This study reports the results from transcriptome profiling revealing that a microtubule associated protein, Ringer (also known as Ringmaker, which is the fly homolog of the mammalian tubulin polymerization-promoting proteins [TPPPs]), is strongly increased following Rtca removal (Monahan Vargas, 2020).
Microtubules and the cytoskeletal network are essential for neuronal function and are paramount to an axon's ability to respond to guidance cues, transport proteins and organelles, grow, survive, and regenerate. Microtubule-binding small molecules and microtubule-associated proteins (MAPs) that regulate microtubule dynamics are attractive therapeutic targets to augment axon regeneration. Ringer belongs to the brain-specific protein, p25α, also known as the TPPP protein family. TPPPs regulate tubulin polymerization and are implicated in neurodegenerative disorders such as α-synucleinopathies and Multiple System Atrophy. Drosophila has only one TPPP ortholog, Ringer, and it directly binds tubulin, promotes microtubule bundling and polymerization in vitro, and is critical for microtubule stabilization and developmental axon growth. This study shows that transcription of ringer is negatively regulated by Rtca via Xbp1. ringer was found to function as a neuronal intrinsic promoter of axon regeneration, working in concert with other MAPs, specifically Futsch/MAP1B and HDAC6, which have been previously shown to be integral for axonal health and integrity. The results reveal MAPs as important arbiters of axon regeneration, and ringer (TPPP homologs) is proposed as an attractive therapeutic target for promoting axon regeneration (Monahan Vargas, 2020).
RBPs have been shown to be crucial in regulating complex cellular processes such as mRNA editing, transport and local translation. Aberrant processing of RNA is present in neuronal diseases and injury. How these processes are affected after nervous system trauma and their regulation during neural repair are poorly understood. Previous work has identified Rtca, an RNA-binding protein regulating RNA repair and splicing, as a potential damage sensor that inhibits axon regeneration. Rtca LOF enhances axon regeneration in both fly and mammalian neurons. To better understand its underlying mechanism, RNA-seq was performed to assess the transcriptome of Rtca mutant neurons; ringer transcripts were found to be highly expressed. Ringer is a MAP homologous to the mammalian tubulin polymerization-promoting proteins (TPPPs), in particular TPPP3 or TPPP1, which has been shown to be a regulator of axonal microtubule organization by promoting microtubule polymerization, assembly, and stability both in vitro and in vivo. This study has revealed a connection between the injury-evoked RNA repair/splicing system and the MAP ringer; it is proposed that Rtca suppresses Xbp1 via nonconventional mRNA splicing, which in turn reduces ringer expression to inhibit axon regeneration. Furthermore, evidence is provided for an association between Futsch and HDAC6, additional MAPs capable of regulating microtubule stability and posttranslational modifications. Ringer is also inhibited by HDAC6, and it cooperates with Futsch to relay a cellular stress signal to the microtubule network. In addition, these data suggest that Rtca and Xbp1 likely have additional downstream effectors independent of ringer, and that Futsch likely receives additional inputs, in parallel to Ringer, to support axonal regeneration. Future studies to directly monitor microtubule dynamics in Rtca LOF mutants will help further validate this model and offer clues to the identity of additional players in this pathway (Monahan Vargas, 2020).
The capacity of an axon to regenerate depends on both the external environment and cell-intrinsic mechanisms, which ultimately converge onto axonal microtubules. MAPs have become popular targets for augmenting nerve regeneration given the importance of microtubule stability and polymerization in both the nascent axon and the regenerating axon's growth cone. As an axon elongates, microtubules engorge the growth cone to fill it with microtubule mass. As the growth cone advances, microtubules bundle and consolidate within the nascent axon to provide structure and support. Ringer has been shown to be essential for proper microtubule bundling. Microtubules are inherently polarized because newly added tubulin dimers only assemble and disassemble at the 'plus' end of the lattice, whereas the minus end of a microtubule is highly stabilized with special tubulin variants, abundant post translational modifications (e.g., acetylation of α-tubulin), and minus-end associating proteins. Therefore, a single microtubule can be thought of as having two general domains; a plus-end that is labile (i.e., where dynamic instability occurs) and a minus end that is stable and resists depolymerization. Microtubule stabilization prevents depolymerization and favors microtubule growth, which is beneficial for the axon's growth cone to advance. Inducing microtubule stabilization using extremely low doses of the drugs paclitaxel or epithilones has resulted in significant augmentation of nerve regeneration in vivo. The results of this study demonstrated a loss of microtubule acetylation in whole-cell lysate and specifically within the proximal axon of injured neurons in ringer mutants. This is in line with the function of Ringer, which has been associated with microtubule polymerization and stability. Future experiments to dynamically track Ringer proteins in accordance with microtubule polymerization during axon regeneration, and an extensive investigation of microtubule posttranslational modifications following axotomy are warranted (Monahan Vargas, 2020).
Futsch, a MAP1B homolog, was recently shown to associate with ringer. Together, Ringer and Futsch were found to regulate synapse formation at neuromuscular junctions via a microtubule-based mechanism. It can be inferred that Ringer and Futsch may help promote the formation of a growth cone rather than a retracting dystrophic end within injured axons, similar to its maintenance of synaptic integrity. Ringer mutation led to a decrease in futsch mRNAs and immunolabeling, suggesting a role in regulating futsch transcription, localization, and protein levels. Both ringer and futsch mutations impaired axon regeneration, albeit futsch had a more dramatic effect, suggesting that futsch may contribute to additional signaling independent of ringer. While heterozygous mutants for futsch and ringer did not have a reduction in regeneration, transheterozygotes of ringer and futsch mutations exhibited a similar reduction in regeneration as ringer mutants alone. Coimmunoprecipitation experiments showed that ringer, futsch, and tubulin physically interact and form a molecular complex, and that Ringer facilitates Futsch binding to tubulin. Epistasis analysis further demonstrated that overexpression of Futsch failed to rescue the reduced axon regeneration in ringer mutants, while overexpression of futsch is sufficient to promote axon regeneration despite the absence of futsch. Importantly, this study found that microtubule turnover is faster in injured versus uninjured axons, and that futsch LOF dysregulates microtubule dynamics, accelerating its turnover after injury. Taken together, sthe data suggest that Ringer and Futsch cooperate in the same complex with tubulin, to maintain microtubule dynamics/stability, and that both are critical to the ability of sensory neurons to regenerate. Futsch is phosphorylated by GSK3 and sustained GSK3 activity promotes axon regeneration and increases the pool of dynamic microtubule mass, which further leads to a speculation that futsch might be regulated by additional signaling pathways (Monahan Vargas, 2020).
Elucidating how microtubule stability properties are altered following an injury and the MAPs responsible for mediating those changes may identify novel therapeutic targets. This study found that acetylation properties were altered by ringer mutations and, therefore, attempts were made to explore the role HDAC6, the primary tubulin deacetylase, may play in instructing regeneration. HDAC6 knockout and pharmacological inhibition increased regeneration in C3da neurons, a subtype of sensory neurons incapable of regeneration in WT flies. Previous studies have shown that HDAC6 inhibition and deletion leads to the hyperacetylation of microtubules. Early studies found that HDAC6 was neuroprotective after a CNS injury and associated these findings with HDAC6's role in transcriptional regulation. However, more recent studies found that HDAC6 is neuroprotective in a manner that was associated with its deacetylation of microtubules. Other studies have shown that HDAC6 is essential for healthy axonal transport and influences MAP-microtubule interactions. This study showd that HDAC6 LOF leads to increased protein levels of ringer and futsch, likely through posttranscriptional mechanisms. It may also be possible that HDAC6 knockout affects microtubule-binding kinetics and the protein localization of Ringer and Futsch (i.e., concentrated versus diffuse). Augmented regeneration following HDAC6 knockout was lost with a ringer mutation. These results, along with the changes observed in Ac-Tub levels, suggest an interaction between HDAC6 and Ringer, where Ringer may function to either directly or indirectly restrict HDAC6 deacetylase activity with respect to α tubulin acetylation. This is likely, given that Ringer has been shown to regulate microtubule bundling and stability, which are associated with highly acetylated domains of microtubules. Ringer may be essential to protecting highly acetylated and stable microtubule domains from HDAC6 deacetylation by occluding its interaction with α tubulin or directly blocking deacetylase activity. This would be consistent with in vitro studies suggesting that mammalian TPPP modulates microtubule acetylation by binding to HDAC6 and inhibiting its activity. Alternatively, HDAC6 could inhibit TPPP nucleation by binding to TPPP and preventing its association to tubulin. Furthermore, HDAC6 can also physically modify kinases shown to negatively interrupt TPPP function such as ERK2. This network hypothesis could help explain an underlying positive feedback loop regulating microtubule stability: Increase of TPPP would inhibit HDAC6 leading to an enhancement of acetylated, potentially stable microtubule; in contrast, modification of kinases by HDAC6 could lead to kinase activation and downstream phosphorylation of TPPP, limiting its microtubule binding activity. It is believed that HDAC6 and ringer are involved in a pathway that ultimately affects the stability and dynamics of microtubules. Future studies will explore whether Ringer and HDAC6 expression, along with posttranslational modifications of tubulin, can predict the regenerative potential of da sensory neurons. C4da neurons show only ~75% regeneration and it is proposed that the other 25% will show differences in the expression of MAPs and microtubule posttranslational modifications, specifically acetylation of α-tubulin (Monahan Vargas, 2020).
The future treatments for nerve regeneration will most likely be combinatorial, with a need to address the extrinsic and intrinsic barriers to regeneration. This study has identified a link between RNA repair/splicing and microtubule organization via a damage-evoked mechanism involving Rtca and Ringer. Further evidence is presented that therapeutic targets capable of augmenting nerve regeneration ultimately converge on microtubules. Microtubules are a bottleneck to regeneration and identifying intrinsic signaling cascades that regulate microtubule dynamics using fly genetics will reveal pathways critical to microtubule-mediated nerve regeneration. Given the complexity of MAPs and the increasing number of candidate proteins, utilizing the fly injury model system allows screening for promising targets that warrant an investigation into their mammalian homologs with in vitro and in vivo mammalian nerve injury models. Excitingly, the zebrafish homolog of TPPP3 was recently shown to promote axon regeneration in Mauthner cells and is regulated at the transcript level by microRNA 133b. This corroborates the current findings, leading to the proposal that ringer/TPPP is tightly regulated and may function as a relay station at multiple levels. Moreover, HDAC6 was also recently shown to be inhibitory in a regeneration screen performed in C. elegans. In summary, this study has identified a RNA repair/splicing pathway that up-regulates the MAP Ringer, which interacts with other MAPs associated with microtubule stability/dynamics and tubulin posttranslational modifications, processes that are evolutionarily conserved and promising targets for regenerative therapies (Monahan Vargas, 2020).
Cortical collapse factors affect microtubule (MT) dynamics at the plasma membrane. They play important roles in neurons. How cortical collapse factors influence axon growth is little understood. This study focussed on the function of Drosophila Efa6 in experimentally and genetically amenable fly neurons. First, it was shown that Drosophila Efa6 can inhibit MTs directly without interacting molecules via an N-terminal 18 amino acid motif (MT elimination domain/MTED) that binds tubulin and inhibits microtubule growth in vitro and cells. If N-terminal MTED-containing fragments are in the cytoplasm they abolish entire microtubule networks of mouse fibroblasts and whole axons of fly neurons. Full-length Efa6 is membrane-attached, hence primarily blocks MTs in the periphery of fibroblasts, and explorative MTs that have left axonal bundles in neurons. Accordingly, loss of Efa6 causes an increase of explorative MTs: in growth cones they enhance axon growth, in axon shafts they cause excessive branching, as well as atrophy through perturbations of MT bundles. Efa6 over-expression causes the opposite phenotypes. Taken together, this work conceptually links molecular and sub-cellular functions of cortical collapse factors to axon growth regulation and reveals new roles in axon branching and in the prevention of axonal atrophy. Furthermore, the MTED delivers a promising tool that can be used to inhibit MTs in a compartmentalised fashion when fusing it to specifically localising protein domains (Qu, 2019).
Axons are the cable-like neuronal extensions that wire the nervous system. They are only 0.1–15 μm in diameter, but can be up to a meter long in humans. It is a fascinating challenge to understand how axons can extend over these enormous distances and branch in orderly manners, but also how these delicate structures can be maintained for a lifetime, that is many decades in humans. It is not surprising that humans gradually lose about 40% of their axons towards old age, and that axon decay is a prominent neurodegenerative phenomenon (Qu, 2019).
Essential for axon biology are the parallel bundles of microtubules (MTs) running all along the axon shaft; these bundles provide (1) structural support, (2) highways for life-sustaining cargo transport, and (3) a source of MTs that can leave these bundles to drive morphogenetic changes. Through being organised in this way, MTs essentially drive processes of axon growth, branching and maintenance. The dynamics of MTs are orchestrated through MT-binding and -regulating proteins, for most of what is known about the molecular mechanisms of function. However, such knowledge alone is usually not sufficient to explain their cellular roles (Qu, 2019).
For example, cortical collapse factors are cell surface-associated proteins which specifically inhibit MTs that approach the cell periphery. Previous reports suggested important roles for cortical collapse factors in regulating axon growth: the ARF activator Efa6 (exchange factor for ARF6) in C. elegans negatively impacts on developmental and regenerative axon growth (Chen, 2015; Chen, 2011; O'Rourk., 2010); the mammalian type four kinesin KIF21A also affects axon growth and links to the neurodevelopmental eye movement disorder 'congenital fibrosis of extraocular muscles' (OMIM reference #135700). However, it can currently only be hypothesized how the molecular functions of these two collapse factors link to axon growth, most likely by acting in growth cones (GCs) (Qu, 2019).
GCs are the amoeboid tip structures through which axons extend to wire the nervous system during development or regeneration. The axonal MT bundles terminate in the centre of GCs; from here, single MTs splay into the actin-rich periphery of GCs. These explorative MTs can trigger extension of the entire MT bundle into their direction, thus elongating the axon; by inhibiting such explorative MTs, cortical collapse factors could negatively impact on axon growth (Qu, 2019).
In line with this argument, and depending on where cortical collapse factors are present and functionally active, further functional predictions could be made: for example, collateral branching of axons along their shafts has been described to depend on explorative MTs that leave the parallel axonal bundles and polymerise towards the periphery. Cortical collapse factors might therefore be negative regulators of axon branching (Qu, 2019).
Other roles might concern axon maintenance: the model of 'local axon homeostasis' states that the force-enriched environment in axons biases MTs to buckle or project out of the bundle to seed pathological areas of MT disorganisation. By inhibiting off-track MTs in the axon shaft, cortical collapse factors might prevent such processes, acting in parallel to other bundle-maintaining factors. For example, spectraplakins serve as spacers that keep polymerising MTs away from the cortex by linking the tips of extending MTs to the axonal surface and guiding them into parallel bundles. Their deficiency in any organism causes severe MT disorganisation, potentially explaining human dystonin-linked HSAN6 ('type six hereditary sensory and autonomic neuropathy'; OMIM ID: 614653). If the hypothesis is correct, loss of cortical collapse factors in axon shafts would also cause MT disorganisation, but through a very different mechanistic route (Qu, 2019).
This study makes use of Drosophila neurons as a well-established, powerful model for studying roles of MT regulators. Using in vitro and cellular assays, this study showed that Drosophila Efa6 is a cortical collapse factor acting through its N-terminal MT elimination domain (MTED). The MTED binds tubulin and blocks MT polymerisation in vitro which indicates that the effect of the peptide is due to a direct interaction between the peptide and tubulin. By localising to neuronal membranes, it only abolishes explorative MTs. This subcellular role translates into negative regulation of axon growth and branching and the prevention of pathological MT disorganisation, both in cultured neurons and in vivo. It is proposed that Efa6 functions as a quality control or axonal maintenance factor that keeps explorative MTs in check, thus playing a complementary role to spectraplakins that prevent MTs from leaving axonal bundles (Qu, 2019).
Axons are the structures that wire the brain and body and are therefore fundamental to nervous system function. To understand how axons are formed during development, can be maintained in a plastic state thereafter, and why they deteriorate in pathological conditions, it is necessary to increase knowledge of axonal cell biology. The MT bundles that form the core of axons are an essential aspect of this cell biology, and understanding how these bundles are regulated and contribute to axon morphogenesis will provide essential insights into axon development and maintenance. This study has addressed fundamental contributions made by cortical collapse factors. This work started from reports that two such factors from distinct protein families both negatively impact on axon growth in species as diverse as C. elegans (CeEfa6) and mouse (Kif21A) (Qu, 2019).
This study found that DmEfa6 likewise acts as a negative regulator of axon growth. Efa6 is a cortical collapse factor, inhibiting MTs primarily via the 18aa long MTED. Since the MTED is the only shared motif with CeEfa6 in an otherwise entirely divergent N-terminus, this clearly demonstrates that the MTED is functionally conserved between both species (Qu, 2019).
Capitalising on Drosophila neurons as a conceptually well-established model for studies of axonal MT regulation, two novel roles were demonstrated for Efa6: as a negative regulator of axon branching and a quality control factor maintaining MT bundle organisation. To perform these functions, Efa6 does not affect the dynamics of MTs contained within the central axonal bundles, but it inhibits mainly those MTs that leave these bundles (see A model for axonal roles of Efa6). By inhibiting explorative MTs in GCs, it negatively impacts on a key event underlying axon growth. By inhibiting off-track MTs in the axon shaft, it tones down the machinery that seeds new interstitial branches, but also prevents these MTs from going astray and causing MT disorganisation (Qu, 2019).
Therefore, this work provides conceptual understanding of cortical collapse factors, which can explain how their molecular functions and subcellular roles in MT regulation link to their reported axonal growth phenotypes during development and regeneration, and to their additional functions in axon branching and maintenance. Apart from existing links of cortical collapse factors to neurodevelopmental disorders, it is therefore predicted that future links will be made to neurodegeneration (Qu, 2019).
During axon growth, MTs constantly polymerise towards the periphery of GCs; the advance of many of these MTs is inhibited at the leading edge, and this work shows that cortical collapse factors are key mediators to this end. Only a fraction of MTs enters filopodia, potentially helped by active guidance mechanisms such as MT-actin cross-linkage (e.g. through spectraplakins, tau, drebrin-EB3). The widely accepted protrusion-engorgement-consolidation model of axon growth proposes that stabilised MTs in filopodia can seed axon elongation events. This model is consistent with the current findings for Efa6. Thus loss of Efa6 can contribute to enhanced axon growth in two ways: firstly, through allowing more MTs to enter filopodia; secondly, by allowing them to dwell in filopodia for longer, thus enhancing the likelihood of their stabilisation. This scenario can explain why loss of Efa6 in C. elegans improves axon re-growth after injury and growth overshoot during development, and why the higher levels of Kif21A levels in GCs causes stalled axon growth (Qu, 2019).
In C. elegans it was shown that axonal injury leads to a re-localisation of CeEfa6 to MT minus ends in the axon core. None of the conditions used in the current study reproduced such behaviour with fly Efa6. Furthermore, it was shown that such central pools of CeEfa6 require their MTED to recruit two kinases: TAC-1 (homologue of TACC/transforming-acidic-coiled-coil) and ZYG-8 (homologue of DCLK/Doublecortin-Like-Kinase). However, in contrast to Efa6, both of these kinases perform growth-enhancing functions and play a secondary, delayed role downstream of Efa6. They are therefore unsuited to explain the direct MT-inhibiting roles of the MTED. In contrast, virtually all structure-function analyses performed with CeEfa6 in developing and regenerating axons perfectly match the data and can be explained through the proposed model. Based on these findings, one might argue that CeEfa6 detachment from the membrane could be the consequence of injury-induced physiological changes that would then pose a threat to axonal MT bundles; localisation to MT minus ends could therefore represent a protective sequestration mechanism. Another C. elegans study reported that loss of Efa6 has no impact on MT length in developing axons, which appears consistent with the current data. They also found an increase in MT numbers, but there is currently no mechanism to explain this in non-injury conditions where CeEfa6 stays at the membrane (Qu, 2019 and references therein).
Interestingly, mammalian Efa6 also plays a role in axon regeneration. However, this mechanism is entirely different, in that it requires the C-terminus to activate Arf6 which, in turn, regulates integrin trafficking at the axon initial segment (Qu, 2019 and references therein).
Axon branching can occur via GC split, in that diverging MTs get stabilised in parallel in the same GC. Alternatively, it can occur through interstitial branching which involves the active generation (e.g. through MT severing) and then stabilisation of off-track MTs. Both models agree with observations in Efa6-deficient/over-expressing neurons: greater/lower numbers of MTs were found in GC and shaft filopodia at 6 hours in vitro, which then correlate with enhanced/reduced axonal branch numbers in mature neurons (Qu, 2019).
If interstitial branch formation is negatively regulated by Efa6, this poses the question as to whether Efa6 has to be actively down-regulated in healthy neurons for branching to occur. Efa6 could either be physically removed from future branch points or its MT inhibition function could be switched off. However, no such regulation appears to be required because Efa6 seems to be in a well-balanced equilibrium. Enough Efa6 appears to be present to inhibit occasional, likely accidental off-track MTs; this capacity is surpassed when the number of off-track MTs is actively increased, for example through MT severing proteins during axonal branch formation. Such a saturation model is supported by experiments with shot: filopodial MT numbers are elevated in shot mutant neurons, although Efa6 is present and functional (as demonstrated by the further increase in filopodial MT numbers in shot Efa6 double-mutant neurons). This is consistent with a model where Efa6 function occurs at a level that is easily saturated when increasing the number of explorative MTs. Such a view would also explain why loss of CeEfa6 promotes axon regeneration in C. elegans, in that the constant base-line of MT inhibition present in the wild-type, is removed in the mutant condition, thus favouring growth-mediating explorative MTs (Qu, 2019).
Axonal MT disorganisation in Efa6-deficient neurons occurs gradually and can even be induced by knock-down of Efa6 at mature stages. Therefore, Efa6 appears to prevent MT disorganisation during axon development and maintenance, as is consistent with its continued expression in the nervous system. Such a continued role makes sense in a scenario where MT bundles remain highly dynamic throughout a neuron's lifetime, constantly undergoing polymerisation to drive renewal processes that prevent senescence (Qu, 2019).
Based on these findings, it is proposed that Efa6 acts as a quality control or maintenance factor within a model of 'local axon homeostasis'. This model states that MTs in the force-enriched environment of axons have a tendency to go off-track and curl up, thus potentially seeding MT disorganisation. Different classes of MT-binding regulators, amongst them spectraplakins, prevent this by actively promoting the bundled conformation. It is proposed that cortical collapse factors act in a complementary way to spectraplakins in that they play no role in maintaining MTs in bundles, but they inhibit those MTs that have escaped the bundling mechanisms (Qu, 2019).
In this scenario, MTs are protected from cortical collapse as long as they are actively maintained in axonal bundles; this can explain the long known conundrum of how axonal MTs extend hundreds of micrometres in relative proximity to the cell cortex in axons, whereas in non-neuronal cells cortical proximity of MTs tends to trigger either their inhibition or tethered stabilisation (Qu, 2019).
This study found that the MTED motif correlates well with MT inhibiting functions of Efa6 family members, whereas the rest of the N-terminus bears no obvious further similarity. Experiments with N-terminal protein and synthetic MTED peptide, both reveal association with MTs/tubulin. The MTED strongly interferes with MT polymerisation. Future co-crystallisation experiments are required to reveal how the MTED works. Given its small size it is hypothesised that it simply blocks assembly, rather than acting via more complex mechanisms such as active promotion of depolymerisation (e.g. kinesin-8 and -13, XMap215) or severing (e.g. spastin, katanin, fidgetin (Qu, 2019).
In any case, the small size of MTEDs might come in handy as experimental tools to inhibit MTs, potentially displaying complementary properties to existing genetic tools such as the kinesin-13 Kif2C, stathmin or spastin. Importantly, the experiments with the CAAX domain have shown that Efa6's MT inhibiting function can be targeted to specific subcellular compartments to clear them of MTs, thus opening up a wide range of future applications (Qu, 2019).
Interestingly, the MT-inhibiting role of Efa6 seems not to be conserved in chordates when taking the MTED as indicator for this function. However, roles of cortical collapse factors in neurons seem to have been taken over by other proteins such as the kinesin-4 family member Kif21A. The CFEOM1-linked Kif21AR954W mutation causes the protein to relocate from the axon shaft to the growth cone of cultured hippocampal neurons. In consequence, increased Kif21A levels in GCs cause reduced axon growth - and this study observed the same with Efa6 over-expression. The decreased levels of Kif21A in proximal axons correlate with a local increase in side branches - and the same is observed with Efa6 loss of function (Qu, 2019).
Finally, this study found that the C-terminal domains of Efa6 might display some degree of functional conservation. So far, work on mammalian PSDs has revealed functions for C-terminal domains in regulating ARF6, ARF1 or ARL14 during actin cytoskeletal reorganisation and membrane ruffling, tumour formation, axon regeneration and immune regulation. The finding that PSD1 and C-terminal Efa6 constructs cause similar membrane ruffling phenotypes in fibroblasts, suggests that some conserved functions reside in this region and might further contribute, together with N-terminally mediated MT inhibition, to the neuronal or non-neuronal defects that cause semi-lethality displayed by Efa6 mutant flies (Qu, 2019).
It is proposed that Efa6 acts as a cortical collapse factor which is important for the regulation of axonal MTs and relevant for axon growth, maintenance and branching. Although this function of Efa6 is evolutionarily not widely conserved, these findings provide a helpful paradigm for studies of other classes of cortical collapse factors also in mammalian neurons. Promising research avenues will be to refine the mechanistic understanding of how Efa6 blocks MT polymerisation, not only to better understand how it can be regulated in axons, but also to better exploit MTEDs as molecular tools in cell biological research (Qu, 2019).
Split ends (SPEN) is the founding member of a well conserved family of nuclear proteins with critical functions in transcriptional regulation and the post-transcriptional processing and nuclear export of transcripts. In animals, the SPEN proteins fall into two size classes that perform either complementary or antagonistic functions in different cellular contexts. This study shows that the two Drosophila representatives of this family, SPEN and Spenito (NITO), regulate metamorphic remodeling of the CCAP/bursicon neurosecretory cells. CCAP/bursicon cell-targeted overexpression of SPEN had no effect on the larval morphology or the pruning back of the CCAP/bursicon cell axons at the onset of metamorphosis. During the subsequent outgrowth phase of metamorphic remodeling, overexpression of either SPEN or NITO strongly inhibited axon extension, axon branching, peripheral neuropeptide accumulation, and soma growth. Cell-targeted loss-of-function alleles for both spen and nito caused similar reductions in axon outgrowth, indicating that the absolute levels of SPEN and NITO activity are critical to support the developmental plasticity of these neurons. Although nito RNAi did not affect SPEN protein levels, the phenotypes produced by SPEN overexpression were suppressed by nito RNAi. It is proposed that SPEN and NITO function additively or synergistically in the CCAP/bursicon neurons to regulate multiple aspects of neurite outgrowth during metamorphic remodeling (Gu, 2017).
The SPEN family has been evolutionarily conserved, with representatives from protists and plants to animals. The family includes mouse MINT (Msx2-interacting nuclear target protein) and human SHARP (SMRT/HDAC1 associated repressor protein). SPEN, SHARP, and MINT are unusually large proteins of ~3575 to 5500 amino acids in length. In addition to SPEN, the Drosophila genome contains one other SPEN-like gene, spenito (nito), that encodes a much smaller, 793 amino acid protein . Although SPEN and NITO share conserved N-terminal RRMs and the SPOC domain, their overall sequence similarity is only 28%, suggesting that SPEN and NITO may function similarly in some processes, but differently in others. For example, SPEN and NITO display functional antagonism during eye development (Jemc, 2006) but act synergistically in regulating Wingless signaling in wing imaginal discs and cultured Kc cells (Chang, 2008). In addition, NITO was identified as a splicing factor involved in Sxl regulation and sex determination in Drosophila, while SPEN had no such effect (Yan, 2015). Several studies in vertebrates also suggest that the relationship between these two proteins is context-dependent (Chang, 2008). Similar to SPEN, NITO is broadly expressed in Drosophila tissues (Chang, 2008). Therefore, this study investigated the function of NITO and its possible interactions with SPEN in the context of remodeling of the CCAP/bursicon neurons (Gu, 2017).
Several previous studies have examined the role of SPEN during neuronal differentiation. In embryos, SPEN contributes to neuronal cell fate specification and regulates the growth, pathfinding, and fasciculation of PNS and CNS axons. SPEN also regulates proliferation and differentiation of Drosophila photoreceptor neurons. In addition to its role in neuronal differentiation, SPEN has also been shown to modify late-onset, progressive neurodegeneration in a model for spinocerebellar ataxia in the mature Drosophila retina. This study reports that SPEN regulates developmental plasticity in mature neurons (Gu, 2017).
SPEN overexpression specifically inhibited neurite outgrowth during metamorphic remodeling of the CCAP/bursicon cells, with little or no effect on the larval morphology of these neurons. The reasons for the stage-dependence of SPEN activity in these cells are unknown, but one intriguing possibility is that the stage-dependence results from a direct or indirect link to the ecdysone titer during metamorphosis. This is suggested by work on the human SPEN ortholog, SHARP. SHARP expression is steroid-inducible. In addition, the RRM domains of SHARP interact directly with the steroid receptor RNA cofactor SRA, which acts as a scaffold to bring together nuclear receptors, corepressors, and coactivators. SHARP inhibits the transcriptional activity of SRA-stimulated estrogen and glucocorticoid receptors. No Drosophila ortholog of SRA has yet been identified. Nevertheless, in future studies, it will be of interest to examine interactions between SPEN and signaling by the ecdysone receptor (EcR) during neuronal remodeling. (Gu, 2017).
The overexpression of spen and nito produced axon growth and branching phenotypesthat were qualitatively similar (although not identical) to the loss-of-function effects of both genes. Such similarities in GOF and LOF phenotypes are often observed in systems where the stoichiometric ratio of gene products to the concentration of other cellular components is important. Thus, the current observations indicate that SPEN may need to be maintained at a specific level, or within an expression window, to properly regulate axonal outgrowth. Recent studies observed strong genetic interactions between spen and multiple factors controlling Myosin II activity. Notably, many cellular movements, including growth cone migration and branching, depend upon dynamic cytoskeletal rearrangements that can be disrupted by either increasing or decreasing the stability or function of cytoskeletal components. Thus, the interaction between SPEN and Myosin II provides one possible explanation for the similar responses of the CCAP/bursicon neurons to reduced versus enhanced levels of SPEN activity. Interestingly, SPEN also interacts genetically with crinkled (Myosin VIIA) in controlling wing vein development and wing bristle positioning, suggesting a more general function of SPEN in cytoskeletal rearrangements. If SPEN and NITO regulate common cellular processes, then this explanation may also apply to NITO (Gu, 2017).
The current results showed that NITO and SPEN act additively or synergistically to regulate CCAP/bursicon cell remodeling. This finding is in agreement with other studies showing that SPEN and NITO are both positive regulators of Wg signaling in developing wing discs and Drosophila Kc cells, and of programmed cell death in the eye disc induced by the pro-apoptotic factors Head involution defective and Reaper. In contrast, SPEN and NITO are antagonistic in regulating photoreceptor number, rhabdomere morphology, and regular ommatidial spacing in the adult eye. Furthermore, SPEN and NITO may play completely different functions in some situations, as NITO regulates Sxl level and its alternative splicing, while SPEN has no such effects (Yan, 2015). Thus, the interaction between SPEN and NITO depends on the cellular context. In mammals, the large and small SPEN-like proteins display similar context-dependence in the regulation of Notch signaling. The large SPEN family proteins MINT and SHARP both suppress Notch signaling by competing with the N intracellular domain (NICD) for binding to the core transcription factor RBP-J. The NITO ortholog, RBM15, also complexes with RBP-J, but it either stimulates or represses expression of a reporter in different cell lines. Therefore, in Drosophila and in vertebrates, large and small SPEN family proteins act redundantly or synergistically in some cellular contexts and antagonistically in others, or they perform completely different roles (Gu, 2017).
Recent insights into the molecular interactions with SPEN family proteins provide some clues to mechanisms underlying these context-dependent differences in function. In different systems, the large SPEN family proteins function as either transcriptional corepressors or coactivators. For example, human SHARP and RBM15 were identified as crucial factors required for the long non-coding RNA Xist-mediated silencing of X-chromosomes by directly interacting with the Xist, recruiting nuclear corepressor, SMRT, activating histone deacetylase3 (HDAC3), and deacetylating histones to exclude Pol II and repress transcription. Other studies have revealed functions of the SPEN proteins in transcription activation. MINT enhances transcriptional activation at the osteocalcin promoter and associates with an actively phosphorylated and processive form of RNA polymerase II. SHARP enhances beta-catenin/T cell factor (TCF)-mediated transcription. In differentiating Drosophila hemocytes, SPEN binds to many target gene promoters in association with a known activating histone modification pattern (Gu, 2017).
The small SPEN proteins have not been shown to function as transcriptional coactivators or corepressors and instead play important roles in alternative splicing, selection of alternative polyadenylation sites, and nuclear export of mRNAs. However, both the large and small SPEN proteins may function together in protein complexes that first associate with nascent transcripts. These messenger ribonucleoprotein (mRNP) complexes are dynamic structures that participate in transcription, pre-mRNA splicing, and nuclear export, and they change in protein composition to permit nuclear export once pre-mRNAs are completely spliced. Importantly, several studies have detected associations among the large and small SPEN proteins in these structures . Given the strong evolutionary conservation of the large and small SPEN proteins, similar interactions are likely to explain some of the overlapping functions of SPEN and NITO during neuronal remodeling in Drosophila (Gu, 2017).
Mutations in motor axon guidance molecules cause aberrant projection patterns of motor nerves. As most studies in Drosophila have analysed these molecules in fixed embryos, the consequences for larval locomotion are entirely unexplored. This study took advantage of sidestep (side)-mutant larvae that display severe locomotion defects because of irreparable innervation errors. Mutations in side affected all motor nerve branches and all body wall regions. Innervation defects were non-stereotypical, showing unique innervation patterns in each hemisegment. Premature activation of Side in muscle precursors abrogated dorsal migration of motor nerves, resulting in larvae with a complete loss of neuromuscular junctions on dorsal-most muscles. High-speed videography showed that these larvae failed to maintain substrate contact and inappropriately raised both head and tail segments above the substrate, resulting in unique 'arching' and 'lifting' phenotypes. These results show that guidance errors in side mutants are maintained throughout larval life and are asymmetrical with respect to the bilateral body axis. Together with similar findings in mice, this study also suggests that miswiring could be an underlying cause of inherited movement disorders (Kinold, 2018).
Precise control of neurite guidance during development is essential to ensure proper formation of neuronal networks and correct function of the central nervous system (CNS). How neuronal projections find their targets to generate appropriate synapses is not entirely understood. Although transcription factors are key molecules during neurogenesis, their entire function during the formation of networks in the CNS is not known. This study used the Drosophila melanogaster optic lobe as a model for understanding neurite guidance during development. The function of Sox102F/SoxD, the unique Drosophila orthologue of the vertebrate SoxD family of transcription factors, was assessed. SoxD is expressed in immature and mature neurons in the larval and adult lobula plate ganglia (one of the optic lobe neuropils), but is absent from glial cells, neural stem cells and progenitors of the lobula plate. SoxD RNAi knockdown in all neurons results in a reduction of the lobula plate neuropil, without affecting neuronal fate. This morphological defect is associated with an impaired optomotor response of adult flies. Moreover, knocking down SoxD only in T4/T5 neuronal types, which control motion vision, affects proper neurite guidance into the medulla and lobula. These findings suggest that SoxD regulates neurite guidance, without affecting neuronal fate (Contreras, 2018).
The Drosophila melanogaster visual system is composed of the retina and the optic lobe, which is divided into four ganglia: lamina, medulla, lobula and lobula plate. The visual inputs travel from the retinal photoreceptors through different optic lobe neurons, where this information is processed, triggering behavioural responses. Correct connectivity between optic lobe neurons is fundamental for sensing visual information. In the past few years, several studies have characterised how transcription factors regulate the development and neuronal composition of the optic lobe. However, while the development of the lamina and medulla has been extensively studied, research has only recently been focused on the development of the lobula complex (lobula and lobula plate) (Contreras, 2018).
The Sox (Sry Box) family of transcription factors is a key regulator of embryonic development. These transcription factors bear a conserved DNA binding domain known as the SRY-related High Mobility Group-box (HMG-box), which was first described in the Sry protein that is fundamental for sex determination in mammals. The Sox family of proteins is subdivided into nine groups, depending on the amino acid composition of their HMG-box. Vertebrate genomes encode approximately 20 members of the Sox family, whereas only eight members have been described in the fruit fly. The Sox transcription factors do not activate or repress gene expression themselves, but act together with partner factors that determine the modulation of target genes (Contreras, 2018).
Sox proteins are important during neural development and different groups of Sox transcription factors are responsible for similar neurodevelopmental processes across species. For instance, SoxB1 group members work in early neurogenesis in vertebrates and invertebrates. The vertebrate SoxB1 protein, Sox2, participates in early events of central and peripheral nervous system development. In a similar manner, the SoxB1 orthologues in Drosophila, SoxNeuro (SoxN) and Dichaete, are also required for proper neurogenesis and the formation of neural stem cells during development. On the other hand, SoxD proteins are generally involved later during nervous system development. Vertebrate Sox5 and Sox6 regulate neural stem cell proliferation, neuronal diversity, neuronal migration and projection formation. Similarly, the Drosophila SoxD orthologue is necessary for the development of the nervous system and loss of SoxD function affects synaptic bouton development at the neuromuscular junction and dendritic arborisation in sensory neurons (Contreras, 2018).
This study analysed the role of the Drosophila melanogaster orthologue of the SoxD family: Sox102F/SoxD during optic lobe development. SoxD is expressed in all optic lobe ganglia and is involved in the morphogenesis of the lobula plate neuropil. RNAi-mediated SoxD knockdown in developing neurons severely alters the morphology of the lobula plate ganglia. These morphological defects are not a consequence of changes in the fate of lobula plate neurons, but result from an alteration in the normal pattern of axon and dendrite formation. Associated with the defects in lobula plate morphology, the fly optomotor response is also impaired upon SoxD downregulation in lobula plate neurons. These results are consistent with the observation that Sox5 is involved in neuronal migration and axon pathfinding in mice, denoting that SoxD function is evolutionary conserved (Contreras, 2018).
According to a phylogenetic analysis, Sox102F has high homology to human Sox5, Sox6 and Sox13 transcription factors; therefore, it is proposed to rename this protein SoxD. During larval brain development, SoxD is expressed in neurons of the medulla and the lobula complex, while in the lamina, SoxD is transiently expressed before LPC differentiation into neurons. SoxD knockdown in all neurons or in lobula plate neurons severely affects the morphology of the lobula plate neuropil, impairing fly optomotor behaviour. Thus, this study shows that SoxD is required for the control of lobula plate T4 and T5 neurite guidance without affecting neuronal fate. Finally, it was demonstrated that misexpression of SoxD is sufficient to alter neurite guidance in photoreceptors and mushroom body Kenyon cells (Contreras, 2018).
The lobula plate is one of the less explored ganglia of the Drosophila visual system. Although neurogenesis has been described in some detail, later stages of development and the mechanisms directing neuronal subtype specification are starting to be described (Pinto-Teixeira, 2018). The lobula plate has an important role in motion detection in insects, while the lobula has a role in the integration of optic stimuli and the behavioural response. Two important neuronal populations in this regard are T4 and T5 neurons, which gather information from the medulla and lobula respectively and make their synaptic outputs in the lobula plate. Upon SoxD knockdown in all neurons (Elav-GAL4), specifically in T4/T5 neurons (IPC-GAL4 plus R42F06-Gal4) or only in T5 neurons (R42H07-GAL4), defects are observed in lobula plate morphology with increasing severity correlated with the number of affected neurons. This result suggests that SoxD may be also required for the developing of other lobula plate neurons and not only for T4/T5 neurons. In addition to RNAi-mediated knockdown experiments, a combination of soxD alleles was used that reduces the gene dosage. This hypomorphic condition showed a similar but milder phenotype, supporting the specificity of this phenotype to SoxD function. Furthermore, analysing SoxD knockdown phenotype in T4 or T5 neurons, alterations were observed in axon/dendrite guidance that could explain the motion perception defects of the adult animal (Contreras, 2018).
Recently, Li (2017) showed that SoxD is involved in neuronal development and degeneration. The Li study demonstrated that SoxD is required for synaptic bouton development at the neuromuscular junction, dendritic arborisation in sensory neurons, olfactory behaviour and climbing. This study describes a similar role for SoxD in the development of the lobula plate, but no evidence was found of apoptosis upon SoxD knockdown in larval stages, suggesting that the phenotype observed was not due to loss of lobula plate neurons but to neurite mistargeting during development. Moreover, it wasshowm that optomotor behaviour is also affected after SoxD knockdown, complementing Li's behavioural observations. This evidence lends support for the role of SoxD in axon and dendrite guidance in different types of neurons of the CNS (Contreras, 2018).
The mechanisms by which lobula plate neurite guidance is controlled by SoxD are unknown. Recently, it was described that Atonal promotes the differentiation of T4 and T5 neurons, while Notch signalling activity discriminates between T4 and T5 neuronal fates. This study observed that loss of SoxD did not affect T4/T5 differentiation markers, suggesting that the function of SoxD lays downstream of the T4/T5 fate decision. Thus, it is proposed that SoxD controls final stages of neuronal differentiation during development (Contreras, 2018).
The overgrowth of neurites observed upon SoxD knockdown in T4 and T5 neurons may result from defects in sensing an inhibitory guidance cue that restrict their growth into the medulla or the lobula. In accordance to this hypothesis, SoxD misexpression strongly affects neurite guidance in at least two different systems: photoreceptors and Kenyon cells. This supports the possible role of SoxD on sensing guidance cues. Additionally, the presence of the synaptic terminal marker in T5 dendrites after SoxD knockdown, suggests problems in neurite differentiation that could contribute to the guidance defects. Interestingly, the R42H07-GAL4 driver used to target T5 neurons was generated using an enhancer from the soxD locus. This enhancer does not recapitulate the entire expression of soxD, which is also expressed in T4 neurons and other lobula plate neurons. However, SoxD knockdown was associated to an increase of the GFP fluorescence driven by R42H07-GAL4, suggesting that SoxD may negatively regulate this enhancer (Contreras, 2018).
The regulation of neurite guidance may not be the only role of Drosophila SoxD during development. Over-expression of SoxD in embryonic neuroblasts and RNAi-mediated knockdown of SoxD in glial cells were reported to severely disrupt embryonic CNS development. Surprisingly, this study did not observe expression of SoxD in glial cells in the larval brain, while it remains unknown whether SoxD is expressed in embryonic glia. Furthermore, SoxD is also relevant for the function of other organs. SoxD knockdown in cardiac cells affects heart anatomy and function, while SoxD-RNAi expressed in wing discs increased the size of the longitudinal veins L2 and L3, and the marginal vein (Contreras, 2018).
Future work should address the signalling pathways upstream of SoxD activation and the SoxD targets that govern the morphogenesis of lobula plate neurons. Interestingly, Sox5 knockout mice show defects in axonal pathfinding of corticothalamic neurons, similar to Drosophila T4/T5 neurons, suggesting a conserved role of SoxD proteins in neurite guidance (Contreras, 2018).
A recent paper depicted the role of Sox5 in the regulation of the Collapsin Response Mediator Protein (CRMP), an intracellular protein involved in neurite guidance. Interestingly the authors showed that Sox5 gain of function reduces neurite guidance through CRMP in hippocampal neurons, shedding some light to the molecular mechanism involved. Future work should address the conservation of this regulation (Contreras, 2018).
Finally, human Sox5 has been implicated in a number of diseases and intellectual disability in humans. Several studies report mutations and deletions in the sox5 locus that are linked to developmental defects. Therefore, using the fly as a model for neurite guidance may be valuable in determining the biological impact of these mutations in the onset of neurological diseases (Contreras, 2018).
In Drosophila, 50 classes of olfactory receptor neurons (ORNs) connect to 50 class-specific and uniquely positioned glomeruli in the antennal lobe. Despite the identification of cell surface receptors regulating axon guidance, how ORN axons sort to form 50 stereotypical glomeruli remains unclear. This study show that the heterophilic cell adhesion proteins, DIPs and Dprs, are expressed in ORNs during glomerular formation. Many ORN classes express a unique combination of DIPs/dprs, with neurons of the same class expressing interacting partners, suggesting a role in class-specific self-adhesion between ORN axons. Analysis of DIP/Dpr expression revealed that ORNs that target neighboring glomeruli have different combinations, and ORNs with very similar DIP/Dpr combinations can project to distant glomeruli in the antennal lobe. DIP/Dpr profiles are dynamic during development and correlate with sensilla type lineage for some ORN classes. Perturbations of DIP/dpr gene function result in local projection defects of ORN axons and glomerular positioning, without altering correct matching of ORNs with their target neurons. The results suggest that context-dependent differential adhesion through DIP/Dpr combinations regulate self-adhesion and sort ORN axons into uniquely positioned glomeruli (Barish, 2018).
The Drosophila olfactory system provides an excellent system to identify these determinants, where neurons belonging to 50 different olfactory receptor neuron (ORN) classes sort out and synapse with their target projection neurons within 50 unique glomeruli in the antennal lobe. Each ORN class is defined by the exclusive expression of typically a single olfactory receptor gene from approximately 80 possibilities in the genome. ORNs of the same class converge their axons into a distinctly positioned and class-specific glomerulus in the antennal lobe, generating 50 unique glomeruli targeted by the 50 different classes of ORNs. The molecular parameters that establish glomerular organization are not known (Barish, 2018).
The organizational logic of the peripheral olfactory system is conserved in many species including mammals. For example, mice have over a million ORNs grouped into ~1000 classes based on the expression of a single OR gene from over 1000 possibilities in the genome. ORNs of the same class converge their axons onto the same glomerulus in the olfactory bulb, where they synapse with 2nd order mitral/tufted cell dendrites. Mammalian olfactory receptors, through ligand-dependent and -independent G-protein coupled signaling, were shown to differentially regulate the expression of CSRs to direct glomerular positioning of ORNs. Drosophila olfactory receptors however, are ligand gated cation channels and do not contribute to glomerular organization. Thus, how the Drosophila olfactory system coordinately positions 50 classes of ORNs into 50 distinct glomeruli requires further study. With its diverse yet workable amount of ORN classes, the availability of the wiring map, and reporters for all ORNs, the Drosophila olfactory system is a powerful model to gain a systems level understanding of how 50 ORN classes can coordinate highly stereotyped organizational patterns in the brain (Barish, 2018).
Adult ORNs in Drosophila are born from the asymmetric division of precursors located in the larval antennal disc, which, during pupal metamorphosis, becomes the adult antenna. ORN axons reach the future antennal lobe by 16-18 hours after puparium formation (APF). No glomeruli can be seen at these stages. Most glomeruli are clearly detectable by antibodies against N-Cadherin, around 40 hours APF, and fully separate into distinct structures by 48-50 hrs APF. Several genes have been shown to regulate each step of ORN axon guidance, such as Semaphorins/Plexins (axonal tract selection, interclass repulsion), Dscam and Robos (axon targeting), N-Cadherin (intraclass attraction), and Teneurins and Tolls (ORN-PN matching). The vast majority of these proteins however, work very broadly and are required for the proper targeting of most or all ORN classes. It is therefore still unclear how axons of 50 different ORN classes organize themselves to form 50 uniquely positioned and structured glomeruli. This is likely due to the complex and combinatorial nature of the molecular interactions underlying ORN wiring patterns, which includes programs for axon-axon sorting/positioning and target specificity in the antennal lobe (Barish, 2018).
This study identified the Defective proboscis response (Dpr) family proteins and their heterophilic binding partners Dpr Interacting Proteins (DIPs) as novel regulators of glomerular positioning and structure in the Drosophila olfactory system. Many ORN classes express a unique combination of DIP/Dprs starting at stages dedicated to glomerular formation. Interestingly, interacting DIP/Dpr partners are generally, but not exclusively, found in the same ORN class, likely aiding self-adhesion among axons of the same ORN class, while also sorting from others. Mathematical analysis of class-specific DIP/Dpr expression showed that ORNs with very similar DIP/Dpr combinations, can end up in distant glomeruli in the antennal lobe, and ORNs targeting neighboring glomeruli can have very different combinations. DIPs/Dprs control the class-specific positioning of ORN axon terminals and their glomerular morphology. Perturbations to DIP/Dpr combinations are associated with context-dependent, and local disruptions of glomerular morphology and positioning and in some cases, invasions of neighboring glomeruli, without changing ORN-PN matching. These results suggest that differential adhesion among local ORN axons, mediated by DIPs/Dprs, determines the position and morphology of each glomerulus. The results demonstrate how combinatorial action of many interacting CSRs can generate differential adhesive forces as a strategy to coordinately organize axons of all circuits within a neural system (Barish, 2018).
This study shows that the Dpr family of Ig-domain transmembrane proteins and their heterophilic binding partners DIPs are expressed in ORN specific combinations. Mathematical analysis of class-specific DIP/Dpr expression profiles suggest ORN classes with similar DIP/Dpr profiles, can target distant glomeruli, and neighboring glomeruli are targeted by ORNs with different DIP/Dpr combinations, suggesting a role in ORN intra-class adhesion and inter-class sorting. The results in vivo are in agreement with this hypothesis, as loss of a single DIP/Dpr gene in a specific ORN class, or multiple genes in all ORNs, causes local disruptions of ORN terminal projections and glomerular positioning, without causing defects in ORN-PN matching. Misexpression of DIPs/dprs causes similar phenotypes, disrupting normal axon-axon interactions in both cell autonomous and non-autonomous ways. Overexpression of DIPs/Dprs in multiple classes of ORNs can cause cell non-autonomous phenotypes in other classes of ORNs, even distant neighbors, suggesting axon-axon interactions can shift glomerular positioning. ORN projection phenotypes in different levels of single protein knock downs, and combinatorial knock down or overexpression of DIPs, consistent with a model where integration of differential adhesive forces by different DIP/Dpr combinations in ORNs contributes to glomerular structure and position. Some Dprs, Dpr10 specifically, also control additional processes during wiring, such as the fate or correct guidance of ORNs to appropriate glomerular regions, as seen in dpr10 mutants. Together, the data reveal that DIPs/Dprs are critical players in ORN axon sorting, as well as the positioning of 50 ORN class-specific glomeruli (Barish, 2018).
Although this study provides a significant advance in understanding how axon terminals for 50 ORN classes segregate into uniquely positioned glomeruli, it is incomplete. First and foremost, expression data is not available for several other DIP/Dpr family members (Dprs 4, 7, 14, 17, 18, 19, 20, 21, and DIPs-α and ι). Second, this study focused on four glomeruli targeted by Or47b, Or88a, Or83c, and Ir84a ORNs, showing only a few examples of manipulations in other ORN classes. More sophisticated systems level genetic analyses of all DIP/Dpr manipulations, in addition to identification of CSR expression profiles in each ORN class and its target projection neuron will help refine this model, in the future (Barish, 2018).
Another caveat to this analysis is that that the ORN class-specific DIP/Dpr profiles rely on MiMIC GAL4 driven reporter expression patterns in the antennal lobes. Even though these have been confirmed in the neurons of the visual system, it is possible that some do not reflect the endogenous gene expression. Thus, acquisition of ORN specific transcriptional profiles, together with targeted GAL4s knock-ins into individual DIP/dpr loci will be needed in the future for a more detailed understanding of the ORN-specific DIP/Dpr combinatorial codes (Barish, 2018).
One key characteristic of the peripheral olfactory system is the sorting of approximately 1500 ORN axon terminals into 50, uniquely positioned, and ORN class-specific glomerular units in the antennal lobe. The data suggest that, in addition to guidance of ORN axons to the antennal lobe and synaptic ORN-PN matching within glomeruli, axon-axon interactions among ORNs also contribute to the glomerular organization and formation. Such axon-axon interaction among ORNs during development can function to attract and adhere ORN axon terminals of the same class, simultaneously sorting from the axon terminals of other ORN classes, which themselves must self-adhere. These interactions can also generate differential forces that also position the terminals of each ORN, and thus the position of each glomerulus with respect to others. The molecular mechanisms driving these different processes during glomerular positioning and formation are not known. The process of sorting subsets of ORN axons into different tracks successively, starts with Notch signaling and its regulation of semaphorins, as ORNs in the same sensillum are born from asymmetric divisions of the same precursor to acquire separate wiring identities. Each of the sibling ORNs take one of the two early axon tracks before glomeruli start to form based on their Notch state, which also determines whether or not they express Sema-2b. Sema-1a signaling also contributes to repulsion of neighboring ORN axons of different classes, but again, effects are relatively general, causing sorting defect of many ORN classes. In addition to repulsive signals, examples of homophilic cell adhesion proteins, such as N-Cadherin, were previously shown to regulate glomerular formation as well, by interfering with axon-axon interactions among the same class of ORNs. The effect of Ncad mutants however, are seen in all glomeruli, which does not explain selective adhesion that occurs among each class. This study shows that ORN-specific combinations of DIP/Dpr pairs regulate glomerular morphology and positioning within the antennal lobe. Knock down and mis-expression experiments also indicate that ORN axons interact via their DIPs/Dpr combinations suggesting that they may distinguish themselves from other ORN axons nearby and integrate these interactions to identify and converge with ORNs of the same class, positioning the glomeruli for other ORNs with similar or slightly compatible DIP/Dpr profiles forming glomerular neighborhoods. This is particularly apparent for trichoid and coeloconic ORNs, which have more similar DIP/Dpr profiles among ORNs within each sensilla type compared to others. Given the previously reported adhesive function of DIP/Dpr interactions (Carrillo, 2015; Tan, 2015), this work then supports a model of differential adhesion that emerges from ORN-specific combinatorial DIP/Dpr profiles as a strategy for class-specific sorting of ORN axon terminals (Barish, 2018).
The sorting defects observed in this study could, in theory, arise due to either defects in intra-class attraction or inter-class repulsion. Taken together, these data argue for a third option, where differential adhesive interactions among axons sort out glomeruli of different classes. In many cases ligand-receptor DIP-Dpr pairs are expressed in the same ORN class, which can interact heterophilically to mediate adhesion, albeit it is also possible that some DIP/Dprs interact homophilically. Yet, some ORN classes, especially ones with more complex combinatorial codes, additionally express only specific dpr ligands, without their DIP receptors. In these cases, the receptors can rather be expressed in ORN classes that target neighboring glomeruli. It is therefore possible that differential axon-axon interactions during development can help position axons from different ORN classes based on its DIP/Dpr profile. Indeed, the results with dpr1, DIP-δ, and DIP-γ overexpression supports this model of interaction. When expressed in only Or47b neurons, these genes yield no significant changes to the Or47b ORN projections or the VA1v glomerulus. Yet only when they are expressed in other ORN classes Or47b projection defects appear. This suggests that during development Dpr1, DIP-Δ, and DIP-γ in Or47b neurons and other ORN classes interact with the matching DIPs or Dprs on nearby axons of other ORN classes. These interactions likely generate forces that pull out Or47b axons to distinct directions. Additional support for differential adhesion comes from the titration levels of DIP-ν knock down, where increasing the temperature and thus the strength of knock down is accompanied by increased severity of the Or47b ORN projection phenotype. This is likely due to differing levels of self-adhesion, where a slight decrease in self adhesion can lead to glomerular splits as opposed to a full expansion of VA1v glomerulus in stronger knock downs to fully overtake the VA1d glomerulus. This is also consistent with the developmental analysis of the phenotype, which reveals that glomerular splits and positional defects are apparent at 45-50hrs APF, before axons fully expand at 55hrs APF. Previous reports have observed that the number of Or47b neurons that are positive for the GFP driven by the Or47b-GAL4 increases over pupal development. This would suggest that the number of Or47b neurons that lack DIP-ν expression at mid-pupal stages are relatively few, resulting in a modest reduction of DIP-ν expression in the population as a whole. This reduction would increase as more neurons express the Or47b-GAL4 resulting in full invasion (Barish, 2018).
Combinatorial knock down and overexpression of DIPs also support a model in which DIP/Dprs mediate differential adhesion. Overexpression of either DIP-δ or DIP-γ on their own, misdirects axon terminals indifferent directions. This is likely due to different context dependent adhesive DIP/Dpr interactions among ORN axons within the antennal lobe glomeruli in each experimental condition. Interestingly, these effects are neutralized when both DIPs are expressed simultaneously, suggesting integration of different adhesive forces exerted on the ORN axon terminals for each class (Barish, 2018).
Heterophilic adhesive interactions among DIPs/Dprs are consistent with their previously reported roles in the Drosophila eye in layer specific matching of photoreceptor cells with their targets in the medulla (Carrillo, 2015). Mutations in DIPs/dprs cause photoreceptors to overshoot their targets because they lack adhesion with their postsynaptic partners. In the current study, ectopic synaptic matching of Or47b ORNs with other PNs were not detected, suggesting DIP/Dprs in ORNs might function in glomerular sorting and positioning, but not ORN-PN matching. However, these experiments are restricted to the analysis of MZ19 reporter, which labels DA1, DC3, and VA1d PNs. Thus, even though it is possible that DIP/Dpr interactions might play a role in ORN-PN matching for other ORN classes, the data is in agreement with a model of differential adhesion as an efficient strategy to form and segregate 50 class specific glomeruli in the antennal lobe. At each stage of the wiring program, axons can interact with their neighbors based on their DIP/Dpr profiles where highest adhesion occurs among the axons of the same ORN class, and perhaps determine the relative glomerular position of other ORN classes nearby. Superimposed onto other earlier regulators of wiring, such as Sema-1a, Sema-2a, and N-Cadherin, glomeruli can be sorted out from one another in a repeatable fashion (Barish, 2018).
The results suggest that loss or addition of DIPs/Dprs generally act locally in a class-specific manner during the last stages of glomerular formation. In many instances, phenotypes can be generated by knocking down or overexpressing DIP/dpr genes using OR-GAL4 drivers, after the onset of olfactory receptor expression. This result is rather interesting given the current view of ORN circuit assembly. For many years, the consensus has been olfactory receptor genes are turned on after the glomerular formation is complete. The results suggest that, at least for olfactory receptors that are turned on early in development, glomerular patterns are not entirely established by the onset of OR expression. The finding that class specific knock down and over expression experiments using Or47b-GAL4 drivers for many DIP genes leads to dramatic defects in the target VA1v glomerulus, indicates ongoing axonal decisions that require DIPs/Dprs after the onset of Or47b expression (Barish, 2018).
In addition, developmental analysis of knock down of DIP-ν in Or47b ORNs shows that glomerular deformation can be detected as early 45 hrs APF during the finalization of glomerular formation. This suggests that DIPs and Dprs play some role in the development of glomerular morphology and positioning, while others may be more critical for the maintenance of proper glomerular shape. This conclusion is bolstered by the observation that some DIPs and Dprs have developmentally dynamic expression patterns, with distinct expression patterns at 40-50 hrs APF, while others show little to no expression at this developmental stage. It is likely that different DIPs and Dprs play different roles in ORN wiring depending on their expression pattern and timing (Barish, 2018).
It should also be noted that some phenotypes arise when other Or-GAL4s, that turn on later, are used in conjunction with the Or47b-GAL4. This suggests that while glomeruli obtain they final shape by 48hrs APF, this morphology is not set and can be altered later in development by the mis-expression or knock down of CSRs (Barish, 2018).
When a specific DIP/dpr gene, or gene combination, is lost or ectopically expressed in ORNs, axons invade the glomerulus targeted by a class of ORNs with the most compatible DIP/Dpr code, and/or which also now has new adhesive properties due to the perturbed genetic state. This role is akin to Dscam, a fellow Ig superfamily protein, involved in controlling dendritic self-avoidance, where combinatorial expression of thousands of dscam splice isoforms regulate recognition of self vs non-self-dendritic processes to produce distinct dendritic zones for each neuron. In the case of DIPs/Dprs in the olfactory system, combinatorial expression of DIPs/dprs regulates sorting of ORN axon terminals that belong to the same class (self) from axon terminals of other ORN classes (non-self) which target neighboring glomeruli in the antennal lobe (Barish, 2018).
How do the class specific patterns of DIP/dpr expression arise? It seems likely that similar mechanisms that lead to the singular expression of a particular olfactory receptor in each neuron would also control the expression of DIPs/dprs. There are three major modes of regulation of OR selection: 1-prepatterning of the antennal disc by transcription factor networks that determine sensilla precursor identity, 2-regulation of neuronal fates by Notch-Delta signaling during asymmetric precursor cell divisions, and 3-terminal selector transcription factors regulating olfactory receptor gene expression and possibly other ORN identifiers. Each mode of regulation clearly controls ORN wiring as prepatterning network mutants change ORN connectivity to converted fates, Notch pathway mutants behave similarly, and factors like Pdm3 regulate glomerular shape as well OR expression. It is likely that each mode of regulation, layered on top of each other, work in concert to control DIP/dpr expression. This would lead to class-specific differences in their DIP/Dpr profiles, which would be generated by the developmental programs mediating terminal differentiation of each ORN class, increasing the complexity of the DIP/Dpr code for each ORN class. There might be some DIP/Dprs, like DIP-ν and dpr12, that are expressed earlier and more abundantly in future ORNs, perhaps starting at precursor stages to coarsely sort early axons. Some DIPs/dprs are indeed expressed in the RNA-seq analysis at 8 hrs APF although at much lower levels when compared to 40 hrs APF and adult antennae. DIP/Dprs expressed in only few classes of ORNs, might be superimposed onto existing DIP/Dpr profiles in later stages of development, as individual ORN identities are defined by the onset of OR expression. Deeper understanding of exactly which transcription factors control DIP/dpr expression and how they relate to larger programs of neuronal specification is needed (Barish, 2018).
The mammalian olfactory bulb is organizationally very similar to the Drosophila antennal lobe. In both organisms, the ORNs that express the same receptor converge their axons onto a single class-specific glomerulus. Mammalian olfactory receptors are G-protein coupled receptors and differentially regulate the expression of adhesive and repulsive CSRs to position and sort ORN specific glomeruli using both ligand dependent and independent signaling. Differential, ligand-independent cAMP signaling from each mammalian olfactory receptor gene regulates graded expression of Semaphorin and Neuropilin in different ORN classes, and control the positioning of each glomerulus. In addition, olfactory receptor neuron activity refines glomerular convergence through differential expression of homophilic adhesion Ig-domain proteins Kirrel2/3, and repulsive transmembrane signaling proteins EphA and EphrinA, which regulate intra-class attraction and inter-class repulsion respectively (Barish, 2018).
In contrast to mammalian olfactory receptors, Drosophila olfactory receptors are ligand gated cation channels, and do not contribute to ORN wiring. Interestingly, DIPs and Dprs share homology with Kirrels and seem to operate with a similar logic. Differential and graded expression of adhesion proteins, Kirrels in mammals and a combination of DIPs/Dprs in flies, mediates class-specific glomerular convergence. They do this by regulating adhesion among the axons from the same ORN class, by creating differential adhesion forces locally in the olfactory bulb based on ORN-specific cell surface receptor profiles. Thus, even though olfactory receptors in mammals and Drosophila are functionally and structurally diverse, this study points to a possible evolutionarily convergent downstream molecular strategies that sort ORN axon terminals into distinct glomeruli in a class specific manner (Barish, 2018).
The confined and crowded environment of developing brains imposes spatial constraints on neuronal cells that have evolved individual and collective strategies to optimize their growth. These include organizing neurons into populations extending their axons to common target territories. How individual axons interact with each other within such populations to optimize innervation is currently unclear and difficult to analyze experimentally in vivo. This study developed a stochastic model of 3D axon growth that takes into account spatial environmental constraints, physical interactions between neighboring axons, and branch formation. This general, predictive and robust model, when fed with parameters estimated on real neurons from the Drosophila brain, enabled the study of the mechanistic principles underlying the growth of axonal populations. First, it provided a novel explanation for the diversity of growth and branching patterns observed in vivo within populations of genetically identical neurons. Second, it uncovered that axon branching could be a strategy optimizing the overall growth of axons competing with others in contexts of high axonal density. The flexibility of this framework will make it possible to investigate the rules underlying axon growth and regeneration in the context of various neuronal populations (Razetti, 2018).
Patterns of synaptic connectivity are remarkably precise and complex. Single-cell RNA sequencing has revealed a vast transcriptional diversity of neurons. Nevertheless, a clear logic underlying the transcriptional control of neuronal connectivity has yet to emerge. This study focused on Drosophila T4/T neurons, a class of closely related neuronal subtypes with different wiring patterns. Eight subtypes of T4/T neurons are defined by combinations of two patterns of dendritic inputs and four patterns of axonal outputs. Single-cell profiling during development revealed distinct transcriptional programs defining each dendrite and axon wiring pattern. These programs were defined by the expression of a few transcription factors and different combinations of cell surface proteins. Gain and loss of function studies provide evidence for independent control of different wiring features. It is proposed that modular transcriptional programs for distinct wiring features are assembled in different combinations to generate diverse patterns of neuronal connectivity (Kurmangaliyev, 2019).
T4/T5 neurons share a common developmental origin, physiological function, and general morphology, but differ in their precise wiring patterns and preferred stimulus. There are eight morphological subtypes of T4/T5 neurons in each column of the lobula plate (LoP) neuropil, comprising the most abundant cell type in the fly visual system. These subtypes can be classified into two quartets of subtypes based on dendritic inputs: the four T4 subtypes share a common set of dendritic inputs in the medulla, and the four T5 subtypes share a different set of dendritic inputs in the lobula (see Single-cell sequencing reveals eight transcriptionally distinct populations of T4/T5 neurons). T4 neurons respond to ON stimuli (i.e. bright edges moving against a dark background) and T5 to OFF stimuli (i.e., dark edges moving across a bright background). T4/T5 neurons can also be classified into four pairs of subtypes (a-d) based on the location of their axon terminals within a given column in layers a-d of the LoP. Each pair responds selectively to visual motion in one of four cardinal directions: posterior, anterior, upwards, and downwards, respectively. Although transcriptional profiling of the adult Drosophila brain revealed a common transcriptional signature for all T4/T5 neurons, genetic programs for individual subtypes have not been identified. This study hypothesized that identification of gene expression programs for individual T4/T5 subtypes during circuit assembly would provide insight into the genetic programs regulating discrete wiring features (Kurmangaliyev, 2019).
This study reports that independent transcriptional programs define the dendritic inputs and axonal outputs of T4/T5 neurons. Gain and loss of function studies indicate that these programs control their corresponding morphological features. These findings suggest that the modular assembly of separate dendritic and axonal transcriptional programs contributes to the diversity of wiring patterns in complex nervous systems (Kurmangaliyev, 2019).
A unique attribute of T4/T5 neurons is that the same dendritic and axonal wiring patterns are reiteratively used among different subtypes; each neuron can be described by a unique combination of one of four types of axonal outputs and one of two types of dendritic inputs. It was anticipated that this property of T4/T5 neurons would provide an opportunity to assess the relationship between specific genetic programs and fundamental features of neuronal architecture (Kurmangaliyev, 2019).
Unsupervised analysis revealed that separable transcriptional programs correlate with these specific wiring features. This study demonstrates through gain and loss of function experiments that these programs control specific axonal targeting features, which are separable from other features (e.g., dendrite targeting). These programs can be re-assembled in a modular fashion to generate neuronal subtypes with different combinations of wiring features. A modular transcriptional architecture may provide a general strategy for discrete modifications to neuronal connectivity in development and evolution (Kurmangaliyev, 2019).
A common T4/T5 neuronal identity is defined by a unique combination of TFs expressed in all subtypes (e.g., Lim1, Drgx, acj6). Perturbation of TFs expressed in all subtypes disrupts overall organization of T4/T5 neurons, including both dendritic and axonal morphologies. This common T4/T5 transcriptional program is further diversified by separable feature-specific transcriptional programs. These programs are defined by three binary (ON/OFF) TF expression patterns, with two TF patterns defining the four axonal outputs and one TF pattern defining the two dendritic inputs. In this way, modular TF codes defining common and feature-specific transcriptional programs give rise to eight T4/T5 subtypes (Kurmangaliyev, 2019).
Four pairs of T4/T5 subtypes with shared axonal outputs (and different dendritic inputs) each target one of four LoP layers, a-d. The ultimate layered architecture of neuropils develops through sequential lamination into increasing numbers of layers. Together with previous results, the findings suggest that the lamination of T4/T5 axonal outputs occurs via two distinct processes, each controlled by a separate TF. Binary expression of bi is required for lamination of the broad a/b from c/d LoP domains (Apitz, 2018), whereas binary expression of grn is required for sublamination of each of these two domains into separate LoP layers. Importantly, perturbation of each TF exclusively disrupts the corresponding lamination step, while not affecting other morphological features of T4/T5 neurons. Similarly, two quartets of subtypes with shared dendritic inputs (and different axonal outputs) were defined by binary expression of TfAP-2. Arborization of dendrites in M10 (T4) or Lo1 (T5) occurs during initial neurite guidance steps, preceding the developmental stages covered in this study. It is hypothesized that differentially expressed genes (DEGs) between T4 and T5 subtypes identified in this analysis contribute to the connections with two distinct sets of presynaptic partners (Kurmangaliyev, 2019).
The binary expression patterns of TFs also mirror the developmental lineages of T4/T5 neurons. a/b and c/d subtypes arise from bi- and bi+ progenitor populations. Neuroblasts from each population undergo two terminal Notch-dependent asymmetric divisions to give rise to the eight subtypes (Pinto-Teixeira, 2018). These divisions correspond to binary expression patterns of grn and TfAP-2, respectively, which act with Notch signaling to regulate wiring. Remarkably, despite divergent developmental trajectories separated by multiple divisions and distinct progenitor pools, all T4 and all T5 subtypes converge onto the same transcriptional programs associated with two types of dendritic inputs. Three regulatory dichotomies could also reflect the evolutionary origin of T4/T5 subtypes and correspond to consecutive duplications of ancestral cell types and circuits (Kurmangaliyev, 2019).
Each axonal and dendritic transcriptional program is characterized by a specific pattern of TFs, as well as a set of cell surface proteins (CSPs), many of which are implicated in regulating wiring in other developmental contexts. These include Ig superfamily proteins in which different paralogs exhibit discrete heterophilic binding specificities, including the beat/side and the dpr/DIP interacting protein families. Interestingly, dynamic expression of these proteins in neurons with shared wiring features was developmentally coordinated. It is envisioned that the synaptic specificity of T4/T5 dendrites and axons are determined by the combined activity of these recognition molecules through interactions with synaptic partners. Future experiments utilizing gain and loss of function analysis, either alone or different combinations, will provide insights into the cellular recognition mechanisms by which synaptic specificity is established (Kurmangaliyev, 2019).
The composite morphological properties of T4/T5 subtypes allowed identification, and thus decouple transcriptional programs for dendrite and axon wiring. Combining separate dendritic and axonal programs, and variations on them, may contribute to the diversification of synaptic specificity in different neuronal subtypes across complex nervous systems (Kurmangaliyev, 2019).
Microtubule polarity in axons and dendrites defines the direction of intracellular transport in neurons. Axons contain arrays of uniformly polarized microtubules with plus-ends facing the tips of the processes (plus-end-out), while dendrites contain microtubules with a minus-end-out orientation. It has been shown that cytoplasmic dynein, targeted to cortical actin, removes minus-end-out microtubules from axons. This study has identified Spindly, a protein known for recruitment of dynein to kinetochores in mitosis, as a key factor required for dynein-dependent microtubule sorting in axons of Drosophila neurons. Depletion of Spindly affects polarity of axonal microtubules in vivo and in primary neuronal cultures. In addition to these defects, depletion of Spindly in neurons causes major collapse of axonal patterning in the third-instar larval brain as well as severe coordination impairment in adult flies. These defects can be fully rescued by full-length Spindly, but not by variants with mutations in its dynein-binding site. Biochemical analysis demonstrated that Spindly binds F-actin, suggesting that Spindly serves as a link between dynein and cortical actin in axons. Therefore, Spindly plays a critical role during neurodevelopment by mediating dynein-driven sorting of axonal microtubules (Del Castillo, 2020).
Several works using different model systems have demonstrated that uniform polarity of microtubules in axons requires activity of cytoplasmic dynein recruited to cortical actin filaments. However, the mechanism that targets dynein to cortical actin remains unknown. In the search for adaptors involved in the recruitment of dynein to F-actin, a targeted RNAi screen was performed, and Spindly, a well-characterized protein that recruits dynein to kinetochores in mitosis, was shown to be required in postmitotic neurons for dynein-dependent organization of microtubules in axons. Depletion of Spindly in Drosophila neurons impairs axonal microtubule sorting; brain of Spindly-depleted third-instar larvae showed severe defects in axonal patterning. These neurodevelopmental defects result in impairment of coordination and locomotion and reduced life span of adult flies. These phenotypes are not caused by reduction of the dynein level or inhibition of dynein-driven organelle transport upon Spindly knockdown. Spindly RNAi defects found in the Drosophila brain are fully rescued by expression of full-length Spindly or the variant deficient in kinetochore binding, but not by variants with mutations in its dynein-binding domain. Together, these data suggest that Spindly plays a role in neurodevelopment through a dynein-dependent pathway (Del Castillo, 2020).
Spindly was originally identified as a mitotic component recruited to the kinetochore in a RZZ-dependent pathway. In mitosis, the formation of stable interactions between kinetochores and dynein in the metaphase plate is required to silence spindle assembly checkpoint, allowing the progression of the cell cycle to anaphase. In Spindly-depleted cells, dynein motors fail to be recruited to the outer plate of the kinetochores, and the lack of stable kinetochore-microtubule contacts results in cell-cycle arrest in metaphase. It is proposed that in postmitotic neurons Spindly is also important for dynein recruitment. However, in the case of neurons, Spindly recruits dynein to the actin cortex in axons. Importantly, two types of experiments show that this postmitotic role of Spindly is independent of its canonical role in mitosis. First, depletion of Rod1, one of the kinetochore components that interacts with Spindly during cell division, did not affect the microtubule polarity. More directly, expression of the Spindly variant lacking its kinetochore-binding domain (Spindly-ΔC) rescues the Spindly RNAi defects in Drosophila neurons. Both observations together suggest that the neuronal Spindly-dependent pathway of dynein recruitment and microtubule organization is different from its canonical mitotic pathway (Del Castillo, 2020).
It has been shown recently that, in addition to its role in cell division, Spindly functions in interphase cells. Both in mammalian cells and in Drosophila, changes of Spindly level negatively impact cell migration. Remarkably, both Spindly and the dynein/dynactin complex are found at the cell cortex at the leading edge of migrating human cells. In good agreement with this observation, biochemical and cellular assays showed that Spindly interacts with F-actin. However, at this point it is unknown whether Spindly interacts with actin directly or whether this interaction is mediated by other proteins. The lack of known/predicted actin-binding domains in Spindly favors the second scenario. It will be very interesting to identify the proteins that form a complex with Spindly in interphase and find components of this complex that are involved in the recruitment of Spindly and dynein to F-actin (Del Castillo, 2020).
Importantly, the loss of dynein activity in Drosophila sensory neurons did not affect microtubule polarity in dendrites, indicating that the microtubule-sorting activity of dynein is restricted to axons. The apparent restriction of the dynein-recruiting Spindly activity to axons is yet to be determined. The data support that Spindly primary localizes in the cell body and axon, although a small fraction of the protein can be found in dendrites. It has been reported that Spindly is posttranslationally modified and that modifications affect its localization and/or functions. For example, farnesylation of Cys602 of human Spindly is essential for Spindly accumulation at prometaphase kinetochores. Spindly can also be phosphorylated by cyclin-dependent kinases during cell division, and S515 of human Spindly is the major phosphorylation site. Interestingly, this modification seems to regulate ZW10 function rather than Spindly localization. It is hypothesized that posttranslational modifications in the amino-terminal domain of Spindly may regulate its role in neurodevelopment. Recently, it has been reported that other kinetochore proteins are important for neurodevelopment. For example, depletion of Mis12, Knl1, and Ndc80 (other kinetochore components) results in abnormal neuromuscular junctions and central nervous system development both in Drosophila and in Caenorhabditis elegans. However, the precise roles of these kinetochore proteins in neurodevelopment are as yet unknown (Del Castillo, 2020).
Is cortical dynein the only factor that sorts microtubule polarity in axons? Data from a number of groups using different model systems support the idea of dynein being the universal motor that sorts axonal microtubules. However, depletion of other proteins also results in microtubule polarity defects in axons. For example, it has been reported that TRIM46, a microtubule-associated protein anchored to the AIS through AnkG, is able to organize uniformly oriented microtubule bundles. Obviously, the S2 screen is not comprehensive, and it is not even possible to exclude that the targets that gave negative results in the S2 screen could in fact be involved in microtubule organization in neurons as S2 cell processes are a very crude model of microtubule organization in neurons. It is likely that the development of a nonpolarized neurite into a fully functional axon is a complex process that requires cooperation of multiple components including dynein, dynein adaptors, other microtubule-binding proteins and components of the AIS. The data shown in this study simply demonstrate that Spindly belongs to this group of proteins and is an important factor in the recruitment of dynein to F-actin. Future work will show how these 'mitotic' components work together to properly organize axonal microtubules (Del Castillo, 2020).
A possible indirect role of RIN in the regulation of cRP mRNAs relates to their upregulation by the Ragulator complex. This study shows that RIN binds mRNAs encoding all five subunits of the Ragulator complex. Thus, positive regulation by RIN of Ragulator subunit synthesis could indirectly promote cRP mRNA translation (Laver, 2020).
Direct and indirect regulation of other aspects of gene expression by RIN are also likely. Potentiation of production of core components of the transcription, splicing, and translation machinery might ensure that none of them becomes rate limiting for gene expression in rapidly developing early embryos. Likewise, adequate ATP production would be ensured by potentiation of expression of mitochondrial ribosomal proteins and components of the electron transport chain (Laver, 2020).
That said, the data suggest that the direct effects of RIN are much more pronounced than its indirect effects. Specifically, this study has shown that, in rin mutants, levels of several target mRNAs are significantly reduced, whereas co-expressed non-targets do not change significantly. It should be noted, however, that these analyses were of a small subset of targets; future global analyses might reveal that indirect targets also change significantly (Laver, 2020).
The data have implications for understanding of how cells respond to stress and the role of SGs in that response. A theme in the cellular stress response is a general downregulation of mRNA expression. For example, stress triggers eukaryotic translation initiation factor 2 subunit alpha (eIF2a) phosphorylation, which prevents translation initiation. This, in turn, triggers polysome disassembly, resulting in SG assembly. The storage of long mRNAs in SGs serves as a further mechanism to downregulate their translation. Based on the current results, it is proposed that the recruitment of RIN/G3BPs into SGs would sequester these proteins from their short target mRNAs in the cytoplasm, serving to downregulate the expression of these transcripts. This could indirectly downregulate global gene expression by limiting the production of proteins involved in translation, transcription, and splicing. Likewise, if RIN/G3BPs serve to upregulate mitochondrial function and ATP production, sequestration could attenuate this aspect of cellular metabolism in stressed cells (Laver, 2020).
The formation and maintenance of microtubules requires their polymerisation, but little is known about how this polymerisation is regulated in cells. Focussing on the essential microtubule bundles in axons of Drosophila and Xenopus neurons, this study showed that the plus-end scaffold Eb1, the polymerase XMAP215/Msps and the lattice-binder Tau co-operate interdependently to promote microtubule polymerisation and bundle organisation during axon development and maintenance. Eb1 and XMAP215/Msps promote each other's localisation at polymerising microtubule plus-ends. Tau outcompetes Eb1-binding along microtubule lattices, thus preventing depletion of Eb1 tip pools. The three factors genetically interact and show shared mutant phenotypes: reductions in axon growth, comet sizes, comet numbers and comet velocities, as well as prominent deterioration of parallel microtubule bundles into disorganised curled conformations. This microtubule curling is caused by Eb1 plus-end depletion which impairs spectraplakin-mediated guidance of extending microtubules into parallel bundles. This demonstration that Eb1, XMAP215/Msps and Tau co-operate during the regulation of microtubule polymerisation and bundle organisation, offers new conceptual explanations for developmental and degenerative axon pathologies (Hahn, 2021).
Neurexins are well known trans-synaptic cell adhesion molecules that are required for proper synaptic development and function across species. Beyond synapse organization and function, little is known about other roles Neurexins might have in the nervous system. This study reports novel phenotypic consequences of mutations in Drosophila neurexin (dnrx), which alters axonal microtubule organization and transport. dnrx mutants display phenotypic similarities with the BMP receptor wishful thinking (wit) and one of the downstream effectors, futsch, which is a known regulator of microtubule organization and stability. dnrx has genetic interactions with wit and futsch. Loss of Dnrx also results in reduced levels of other downstream effectors of BMP signaling, phosphorylated-Mad and Trio. Interestingly, postsynaptic overexpression of the BMP ligand, Glass bottom boat, in dnrx mutants partially rescues the axonal transport defects but not the synapse undergrowth at the neuromuscular junctions. These data suggest that Dnrx and BMP signaling are involved in many diverse functions and that regulation of axonal MT organization and transport might be distinct from regulation of synaptic growth in dnrx mutants. Together, this work uncovers a novel function of Drosophila Neurexin and may provide insights into functions of Neurexins in vertebrates (Banerjee, 2018).
Synapse formation, maturation, and turnover require a finely regulated transport system that delivers selected cargos to specific synapses. However, the supporting mechanisms of this process are not fully understood. The present study unravels a new molecular system for vesicle-based axonal transport of proteins in male and female flies (Drosophila melanogaster). This study identifies the gene CG14579 as the transcription unit corresponding to the regulatory mutations known as central complex broad, ccb. These mutations were previously isolated for their morphological phenotype in R-neurons of the ellipsoid body, a component of the central complex. Mutant axons from R-neurons fail to cross the midline, which is indicative of an aberrant composition of the growth cone. However, the molecular mechanism remained to be deciphered. This study shows that CCB is involved in axonal trafficking of FasII and Synaptobrevin, but not Syntaxin. These results suggest that axonal transport of certain proteins is required for the correct pathfinding of R-neurons. The molecular network supporting the CCB system was further investigated, and CCB was found to co-localize and co-immunoprecipitate with Rab11. Epistasis studies indicated that Rab11 is positioned downstream of CCB within this axonal transport system. Interestingly, ccb also interacts with Actin and the Actin nucleator Spire. The data revealed that this interaction plays a key role in the development of axonal connections within the ellipsoid body. It is proposed that the CCB/Rab11/SPIRE system regulates axonal trafficking of synaptic proteins required for proper connectivity and synaptic function (Martin-Pena, 2019).
The actin cytoskeleton drives cell motility and is essential for neuronal development and function. LIM and SH3 Protein 1 (LASP1) is a unique actin-binding protein that is expressed in a wide range of cells including neurons, but its roles in cellular motility and neuronal development are not well understood. LASP1 is expressed in rat hippocampus early in development, and this expression is maintained through adulthood. High-resolution imaging reveals that LASP1 is selectively concentrated at the leading edge of lamellipodia in migrating cells and axonal growth cones. This local enrichment of LASP1 is dynamically associated with the protrusive activity of lamellipodia, depends on the barbed ends of actin filaments, and requires both the LIM domain and nebulin repeats of LASP1. Knockdown of LASP1 in cultured rat hippocampal neurons results in a substantial reduction in axonal outgrowth and arborization. Finally, loss of the Drosophila homolog Lasp from a subset of commissural neurons in the developing ventral nerve cord produces defasciculated axon bundles that do not reach their targets. Together, these data support a novel role for LASP1 in actin-based lamellipodial protrusion and establish LASP1 as a positive regulator of both in vitro and in vivo axon development (Pollitt, 2020).
RAC1 is a highly conserved Rho GTPase critical for several cellular and developmental processes. De novo missense RAC1 variants cause a highly variable neurodevelopmental disorder. Most previously reported patients with this disorder have either severe microcephaly or severe macrocephaly. This study describes eight patients with pathogenic missense RAC1 variants affecting residues between Q61 and R68 within the switch II region of RAC1. These patients display variable combinations of developmental delay, intellectual disability, brain anomalies such as polymicrogyria, and cardiovascular defects with normocephaly or relatively milder micro- or macrocephaly. Pulldown assays, NIH3T3 fibroblasts spreading assays and staining for activated PAK1/2/3 and WAVE2 suggest that these variants increase RAC1 activity and over-activate downstream signalling targets. Axons of neurons isolated from Drosophila embryos expressing the most common of the activating variants are significantly shorter, with an increased density of filopodial protrusions. In vivo, these embryos exhibit frequent defects in axonal organization. Class IV dendritic arborisation neurons expressing this variant exhibit a significant reduction in the total area of the dendritic arbour, increased branching and failure of self-avoidance. RNAi knock down of the WAVE regulatory complex component Cyfip significantly rescues these morphological defects. These results establish that activating substitutions affecting residues Q61-R68 within the switch II region of RAC1 cause developmental syndrome (Banka, 2022).
An 'interactome' screen of all Drosophila cell-surface and secreted proteins containing immunoglobulin superfamily (IgSF) domains discovered a network formed by paralogs of Beaten Path (Beat) and Sidestep (Side), a ligand-receptor pair that is central to motor axon guidance. This study describes a new method for interactome screening, the Bio-Plex Interactome Assay (BPIA), which allows identification of many interactions in a single sample. Using the BPIA, four more members of the Beat-Side network were 'deorphanized'. Interactions were confirmed using surface plasmon resonance. The expression patterns of beat and side genes suggest that Beats are neuronal receptors for Sides expressed on peripheral tissues. side-VI is expressed in muscle fibers targeted by the ISNb nerve, as well as at growth cone choice points and synaptic targets for the ISN and TN nerves. beat-V genes, encoding Side-VI receptors, are expressed in ISNb and ISN motor neurons (Li, 2017).
Protein-protein interactions (PPIs) control a vast array of processes in metazoans, ranging from signal transduction and gene regulation within cells to signaling between cells via cell surface and secreted proteins (CSSPs). The strength of PPIs varies widely, from high-affinity interactions that create stable protein complexes to weak transient interactions. Defining global PPI patterns ('interactomes') has been the focus of much recent research. Progress has been made in generating high-throughput protein interaction data for a variety of organisms, including S. cerevisiae, C. elegans and D. melanogaster. Methods used to create interactomes include affinity purification/mass spectrometry (AP-MS) and the yeast two-hybrid assay (Y2H) (Li, 2017).
It is estimated that up to one sixth of human genes encode CSSPs. CSSPs control signaling from the extracellular milieu to cells and the flow of information between cells. Due to their importance and accessibility, CSSPs are often the targets for therapeutic agents, including humanized monoclonal antibody drugs such as checkpoint inhibitors, the non-Hodgkin's lymphoma drug Rituxan, and the breast cancer drug Herceptin. However, the biochemical properties of many CSSP interactions prevent them from being detected by commonly used techniques employed in high-throughput PPI screens, and CSSPs are underrepresented in global interactomes. There are several reasons for this. First, these proteins are usually glycosylated and have disulfide bonds, so they need to be expressed in the extracellular compartment. CSSP interactions between monomers are also often weak, with KDs in the μM range, making them difficult to capture due to their short half-lives. Lastly, insoluble transmembrane domains on cell surface proteins preclude their purification with standard biochemical techniques, which makes them difficult to study using methods such as AP-MS (Li, 2017).
Despite these difficulties, recent advances have been made in the study of global CSSP interaction patterns. Interactions among cell-surface proteins (CSPs) often occur between clusters of proteins on cell surfaces, and avidity effects (stronger binding due to clustering) can make these cell-cell interactions stable even when monomers bind only weakly. To facilitate detection of interactions among CSSP extracellular domains (ECDs) in vitro, several groups have taken advantage of avidity effects by attaching ECDs to protein multimerization domains and expressing ECD fusions as soluble secreted proteins. These methods have been shown to be effective, allowing detection of interactions that otherwise would not have been observable (Li, 2017).
Özkan (2013) scaled up the avidity-based approach, developing a high-throughput ELISA-like screening method, the Extracellular Interactome Assay (ECIA). The ECIA was used to define interactions among 202 Drosophila CSSPs, comprising all CSSPs within three ECD families. These were the immunoglobulin superfamily (IgSF), fibronectin type III (FNIII) and leucine-rich repeat (LRR) families. The ECIA utilized dimers as 'bait' and pentamers as 'prey'. It detected 106 interactions, 83 of which were previously unknown (Li, 2017).
The most striking finding from the ECIA interactome was that a subfamily of 21 2-IgSF domain CSPs, the Dprs, selectively interacts with a subfamily of 9 3-IgSF domain CSPs, the DIPs, forming a network called the 'Dpr-ome' (Özkan, 2013). Each Dpr and DIP that has been examined is expressed by a small and unique subset of neurons at each stage of development. One Dpr-DIP pair is required for normal synaptogenesis and influences neuronal cell fate. In the pupal optic lobe, neurons expressing a Dpr are often presynaptic to neurons expressing a DIP to which that Dpr binds in vitro. The Dpr-ome initially defined by the global interactome contained several 'orphans', proteins with no binding partner (Özkan, 2013). By expressing new versions of Dprs and DIPs, including chimeras, and using these to conduct a 'mini-interactome' analysis of the Dpr-ome, it was possible to find partners for all but one orphan. That protein, Dpr18, has changes to conserved binding interface residues and may lack the capacity to bind to any DIPs (Li, 2017).
The ECIA also identified a second IgSF network, formed among members of the Beaten Path (Beat) and Sidestep (Side) protein subfamilies. Beat-Ia and Side were identified by genetic screens for motor axon defects, and were later shown to have a ligand-receptor relationship. They control the projection of motor axons to muscle targets. Beat-Ia is expressed on motor axons, where it binds to Side, which is expressed on muscles. This binding causes motor axons to decrease their adhesion to each other, allowing them to leave their bundles and turn onto the muscle fibers. beat-Ia and side have strong motor axon phenotypes. In the absence of either protein, motor axons often remain in their fascicles and never leave to arborize on their target muscles (Li, 2017).
The ECIA detected the known Beat-Ia::Side interaction, and also uncovered other interactions between members of the Beat and Side subfamilies. Seven of the 14 Beats were found to bind to four of the eight Sides. The remaining Beats and Sides were still orphans with no binding partners in the other subfamily. The functions of the newly defined interactions between Beats and Sides were unknown. Most beat genes are expressed in embryonic neurons. Some Beats were genetically characterized using deletion mutations and RNAi, but loss of these Beats did not cause strong motor axon phenotypes. None of the other Side subfamily members had been examined (Li, 2017).
This paper describes the development of a new method for interactome screening, which is called the BPIA (Bio-Plex Interactome Assay). This method uses the 'Bio-Plex' system, which employs Luminex xMAP technology. This method detects binding of a prey protein to many bait proteins, each conjugated to a bead of a different color, in each assay well. For the ECIA, the number of assays required for the interactome screen was the square of the number of proteins examined, while with the Bio-Plex the number of assays could be equal to the number of proteins. In principle, then, the Bio-Plex might greatly speed up interactome screening, and might also be more sensitive, since the available signal-to-background ratio is much greater for the Bio-Plex than for the ECIA. As a test of the method, a Bio-Plex 200 was used to do a mini-interactome screen of the Beat-Side network. Based on the the fact that the Dprs and DIPs that were initially orphans were later shown to have binding partners, it was hypothesized that some of the orphan Beats should have Side partners, and vice versa. Consistent with this hypothesis, it was possible to deorphanize two more Beats and two Sides using the BPIA (Li, 2017).
To further understanding of Beat and Side function during embryonic development, this study characterized expression of several Beats and Sides, focusing primarily on Side-VI and the three Beat-Vs, which were the strongest interactors in both the ECIA and BPIA screens. The three beat-Vs exhibit differential expression in identified motor neurons, while side-VI is expressed at motor axon choice points and in a subset of target muscle fibers (Li, 2017).
In principle, the Bio-Plex system can allow 500 unique protein-protein (bait-prey) interaction pairs to be analyzed in a single well. In this method, capture of proteins from media with streptavidin-coupled beads bypasses the purification step for bait proteins. The assay is also compatible with the use of unpurified prey proteins, thereby reducing the workload for multiplexed screenings. The small size of the beads, the ability to probe multiple interactions simultaneously, and the small volume of the binding reactions all help reduce the amount of protein and reagents needed for the assay. It was possible to produce enough bait and prey proteins for the mini-interactome described in this study (a 23 x 23 matrix) with a single 10 cm dish transfection per protein (Li, 2017).
As a test of the system, the BPIA was used to examine interactions between the Drosophila Beat and Side IgSF protein subfamilies. Beat-Ia is a receptor on motor growth cones that recognizes Side expressed on muscles, and in the absence of Beat-Ia or Side motor axons fail to leave their bundles and arborize on their muscle targets. There are 14 Beat subfamily members and 8 Side subfamily members, but all of these proteins except Beat-Ia and Side itself were orphans until the global IgSF interactome revealed interactions between six other Beats and three Sides (Li, 2017).
In the Dpr-ome, the other IgSF network uncovered by the interactome, every Dpr protein likely to be capable of binding has an interaction partner in the DIP subfamily. Based on this, it is predicted that there should be additional interactions to be discovered within the Beat-Side subfamily network. Using the BPIA, three new interactions were found: Beat-VI::Side-II, Beat-Ic::Side-III, and Beat-Ic::Side. These results suggest that the BPIA is more sensitive than the ECIA. Like the ECIA, the BPIA should be able to find new receptor-ligand interactions even if proteins not previously known to have any interactions were tested. Of course, for both assays any candidate receptor-ligand pairs need to be confirmed as genuine using other methods. For the Beat-Side network, all three new interactions found by the BPIA, as well as the interactions between the three Beat-Vs and Side-VI found by the ECIA, were verified by SPR. This study also demonstrated that Beat-Vs and Side-VI interact using cell-based binding assays and binding to live-dissected embryos (Li, 2017).
There are still five Beats and two Sides that remain orphans. Since the structure of Beat-Side complexes is unknown, it cannot be determined whether these Beats and Sides are likely to be able to bind, but it is speculated that at least the three Beat-IIIs are likely to have Side partners. The Beat-II and Beat-V clusters each interact with a single Side partner, and perhaps the Beat-IIIs interact with one of the two orphan Sides. It is possible that changes in methodology, such as using more highly multimerized preys and/or baits, could increase sensitivity and allow detection of additional interactions (Li, 2017).
The expression patterns of side and beat genes were examined in order to obtain insights into their possible functions. Most sides are expressed in cells in the periphery as well as in the CNS, while most beats are expressed only by CNS neurons, including motor neurons. Beat-Ia::Side interactions are required for normal motor axon guidance, and highly penetrant motor axon defects in which muscles remain uninnervated are observed in mutants lacking either protein. By contrast, partial loss of function of beat-Ib, beat-Ic, both beat-IIs, or beat-VI causes motor axon defects with less than 20% penetrance. Genetic redundancy is a common theme in motor axon guidance , so it is not surprising that only low-penetrance defects are observed when Beat paralogs are not expressed. Given that Beat-Ia and Side both interact with other partners, it is perhaps remarkable that beat-1a and side have such strong phenotypes as single mutants (Li, 2017).
Beat-V and Side-VI also have redundant functions in motor axon guidance. side-VI is expressed in motor axon targets, including muscles 12 and 13 and interacts with the three Beat-Vs, at least two of which are expressed in subsets of motor neurons. Beat-V::Side-VI interactions produced the strongest signals in both the ECIA and BPIA. Sow-penetrance (~1/5 of stage 17 embryonic hemisegments affected) muscle 12 innervation defects were observed in side-VI insertion mutants or in deletion mutants lacking all three beat-V genes. There were also low-penetrance ISN guidance defects in both mutants. The fact that most muscle 12 s are innervated normally in beat-V or side-VI mutants indicates that, while Beat-V::Side-VI interactions may contribute to correct targeting of the RP5 axon to muscle 12, other cues must also be involved. Muscles 12 and/or 13 also express Wnt-4 (a repulsive ligand) and the LRR protein Capricious (Caps; probably an adhesion molecule), and low-penetrance RP5 targeting defects are observed in Wnt-4 and caps mutants. Perhaps muscle 12 is distinguished from other nearby muscles by a set of partially redundant cues, so that strong targeting phenotypes are not observed in any single mutant (Li, 2017).
Although Beat and Side paralogs may not be central to motor axon guidance, their expression patterns suggest that they could be important for determining synaptic connections within the CNS. side-VIII, encoding an orphan Side, is expressed in a small subset of embryonic CNS neurons. In the optic lobe of the pupal brain, an RNAseq analysis of two photoreceptors (R7 and R8) and five types of lamina neurons (L1-L5) revealed that beats and sides have highly specific expression patterns. For example, beat-VII is specific to L2, beat-VI to L5, beat-IIa to L3 (with lower levels in L4), and beat-IIIc to R8, being expressed at much higher levels in those cells relative to all other cells. side and side-III are specific to L3, side-II is specific to L1, side-IV is specific to L2, and side-V is specific to L5. Each of the L neuron types as well as R7 and R8 synapse with different sets of neurons in the medulla, a ten-layered structure that processes visual information from the retina and lamina. It has been observed that R and L neurons expressing specific Dprs often form synapses on medulla neurons expressing DIPs to which those Dprs bind in vitro. In a similar manner, perhaps some of the medulla neurons that are postsynaptic to L or R neurons expressing specific Sides or Beats express their in vitro binding partners, and these Beat-Side interactions might be important for synapse formation or maintenance (Li, 2017).
Despite several decades of studies on the neuromuscular system, the relationship between muscle stem cells and motor neurons remains elusive. Using the Drosophila model, evidence is provided that adult muscle precursors (AMPs), the Drosophila muscle stem cells, interact with the motor axons during embryogenesis. AMPs not only hold the capacity to attract the navigating intersegmental (ISN) and segmental a (SNa) nerve branches, but are also mandatory to the innervation of muscles in the lateral field. This so-far-ignored AMP role involves their filopodia-based interactions with nerve growth cones. In parallel, the expression of the guidance molecule-encoding genes sidestep and side IV in AMPs is reported. Altogether, these data support the view that Drosophila muscle stem cells represent spatial landmarks for navigating motor neurons and reveal that their positioning is crucial for the muscles innervation in the lateral region. Furthermore, AMPs and motor axons are interdependent, as the genetic ablation of SNa leads to a specific loss of SNa-associated lateral AMPs (Lavergne, 2020).
During Drosophila embryogenesis, a stereotypical pattern of AMPs per abdominal hemisegment in ventral (V-AMP), lateral (L-AMPs), dorsolateral (DL-AMPs) and dorsal (D-AMPs) positions can be distinguished. This study has investigated the relationship between AMPs and motor axons, and their dynamics, during development using embryos carrying the M6-gapGFP transgene, which allows visualization of the membrane of AMP cells. The intersegmental nerve (ISN) established contacts with the DL-AMPs during the embryonic stage 13 and then navigated toward the D-AMP to contact it at stage 15. Within the lateral field, the segmental nerve a (SNa) is sub-divided into two branches, dorsal (D-SNa), which innervates the lateral transverse muscles (LTs 1-4), and lateral (L-SNa), which targets the segmental border muscle (SBM). The SNa sub-division takes place during stage 15 and it was observed that the L-SNa branch migrated towards the L-AMPs before innervating the SBM. In parallel, the anterior L-AMP underwent shape changes and directional migration towards the L-SNa. In a similar way, one of the DL-AMPs moves dorsally following ISN migration and the D-AMPs appear to extend toward the ISN. However, AMPs survival and behavior are not affected in the absence of motor axons, as shown in the prospero mutant, where motor axons fail to exit the CNS. To better characterize dynamics of AMP-motor axons interactions, the M6-GAL4; UAS-Life-actin GFP reporter line was used that allows in vivo visualization of both the motor axons and the AMPs. The M6-Gal4 and M6-gapGFP lines are both driven by the same regulatory elements; however, the expression in motor axons, which is low and difficult to distinguish in M6-gapGFP embryos, is enhanced by the GAL4/UAS system and is clearly present in the M6>lifeActGFP context. Live imaging revealed that, among the numerous oriented cytoplasmic projections sent out by the AMPs, those contacting the growth cones of motor axons became stabilized. In particular, stabilization of filopodial connections between L-AMPs and SNa coincided with the setting of the SNa branching point and specification of its lateral branch. Oriented filopodial dynamics were found in the dorsal region with the contact between D-AMP projections and ISN growth cone prior to ISN migration toward the D-AMP. As previously demonstrated, muscle founders are needed for terminal defasciculation of the main motor axon branches. In this context, AMP positioning and the fact that they actively engage with the navigating motor axons might also participate in this process by acting as spatial check-points that either induce and or attract targeted defasciculation of ISN and SNa (Lavergne, 2020).
To investigate the impact of L-AMPs on the SNa pathway and branching, the effect of a genetic ablation of the AMP cells was assessed using the M6-GAL4-driven expression of the pro-apoptotic gene reaper. This enabled targeted induction of apoptosis in AMPs, leading to AMP cell loss without strong defects in the ISN and SNa trajectory, despite the expression of M6-GAL4 in the motor neurons. This differential effect could be due to a lower expression level of M6-GAL4 in motor neurons than in AMPs, and/or a stronger resistance of neural cells to the Reaper-induced apoptosis. Importantly, in 86% of hemisegments, complete loss of L-AMPs was associated with absence of the lateral branch of SNa (L-SNa), strongly suggesting that L-AMPs play an instructive role in L-SNa formation and/or stabilization. In contrast, loss of L-SNa in hemisegments where the L-AMPs were still present occurred in 5.6% of analyzed hemisegments. The L-SNa loss in this context was thus higher than the one observed in the M6-GAL4 line with only 1.8% of hemisegments without L-SNa. To explain this difference, an effect of Reaper expression in the motor system cannot be excluded, but this could also be a consequence of early stages of apoptosis in L-AMPs. Thus, M6-GAL4-induced apoptosis created a context in which loss of the L-SNa branch was observed in L-AMP-devoid segments where the L-SNa target muscle (SBM) was still present. This suggests that L-SNa branch formation might not be dependent on its muscle target, and so prompted a test of whether the L-SNa would form or persist in an SBM-devoid context. reaper expression was targeted to the developing SBM using the SBM(lbl)-GAL4 driver. The SBM(lbl)-GAL4-driven apoptosis resulted in a systematic loss of the SBM and only sporadic loss of the L-AMPs (12% hemisegments). In the SBM-devoid context but with L-AMP cells correctly located, the L-SNa branch was still present (73% of hemisegments). Additionally, in a subset of SBM-deficient embryos, L-AMPs shifted toward the navigating SNa, leading to a shortened L-SNa (13% of the hemisegments). These observations thus suggest an instructive role for AMPs in L-SNa establishment, and reveal that SBM might not be needed for this process and is at least dispensable for its stabilization. To further test the role of L-AMPs in lateral defasciculation of the SNa, different genetic contexts were analyzed in which AMP specification is affected. First a perturbation of asymmetric cell divisions was induced. To adversely affect divisions of progenitor cells that give rise to AMPs, the asymmetry determinant Numb was ectopically expressed using the pan-mesodermal driver Twist-GAL4. In the lateral region, this led predominantly to the loss of one of the L-AMPs and a duplication of the SBM with no major impact on L-SNa formation compared with the control Twist-GAL4 line. However, in a small subset of hemisegments, loss of both L-AMPs but not SBM (often duplicated) was observed. In this rare context, the L-SNa was absent in 88% of hemisegments, supporting the view that L-AMPs are required for L-SNa branching. These findings are also consistent with the effects of generalized mesodermal expression of the identity gene Pox meso (Poxm), which can lead to a loss of L-AMPs without affecting SBM. In such a context, the L-SNa formation is impaired in 89% of L-AMP-devoid segments against 37% in random Twi>Poxm hemisegments. Interestingly, pan-mesodermal expression of Pox meso can also induce misplacement of L-AMPs along the SBM, leading to aberrant L-SNa trajectory. Hence, L-AMPs and their spatial positioning appear crucial to achieve the formation and correct pathfinding of the L-SNa (Lavergne, 2020).
The findings described above suggest that L-AMPs are a source of attractive signals that promote lateral sub-branching of the SNa, making it competent to innervate the SBM. Interestingly, the SBM is the only lateral muscle innervated by the Connectin-positive SNa, which does not express this homophilic target recognition molecule. In such a context, L-AMP-mediated lateral sub-branching of SNa offers a way to drive L-SNa to its specific muscle target. As L-AMPs seem not to express Connectin either, in contrast to previous suggestions, their role in attracting SNa and inducing the L-SNa sub-branching might rely on other guidance molecules (Lavergne, 2020).
It has been previously shown that the mutants of sidestep and beat-1a, which encode interacting membrane proteins of the immunoglobulin superfamily, displayed loss of L-SNa, a phenotype similar to the one observed when L-AMPs are missing. However, the mechanisms leading to the loss of the L-SNa in sidestep and beat-1a mutants have not been elucidated. In addition, the embryonic expression pattern of sidestep has been only partially characterized. By using in situ hybridization, this study found that sidestep mRNA is strongly enriched in all the AMPs, suggesting its potential involvement in the dialogue between AMPs and motor axons. It was therefore decided to test Sidestep protein distribution at the time when L-SNa sub-branching is taking place. By examining stage 14 to 15 embryos, a previously unreported faint and transient expression of Sidestep was found specifically in L-AMPs. To confirm this observation, the expression of Sidestep was analyzed in a mutant for beat-1a. It has been reportedthat the contact of Beat-1a-expressing motor axons with Sidestep-expressing cells leads to a negative regulation of the expression of sidestep. If this contact is missing, cells normally expressing sidestep transiently and at a low level will continue to do so, leading to continuous and higher Sidestep level in these cells. Analyses of beat-1a mutants confirmed that the L-AMPs are Sidestep-expressing cells and that Sidestep expression onset coincides with L-SNa sub-branching. The high Sidestep expression resulting from the lack of beat-1a was still detected in late-stage embryos in which it became gradually restricted to the most anterior L-AMPs. This late differential Sidestep expression may point to a leading role for the anterior L-AMPs in the process of interaction with SNa and in its lateral sub-branching. Additionally, an increased Sidestep expression in L-AMPs was also observed in SNa-devoid pros mutants and in the Duf-GAL4; UAS-NetrinB (NetB) context. Importantly, the SBM does not express Sidestep, making the L-AMPs the only Sidestep-expressing cells in the L-SNa pathway. Thus, this newly reported expression pattern suggests that L-AMPs could attract the L-SNa through the temporally and spatially restricted expression of sidestep (Lavergne, 2020).
Interestingly, the sidestep mutants also display a stall phenotype of the ISN suggestive of a potential role of the D-AMPs. Indeed, this study observed that the aberrantly located D-AMPs, after the mesodermal overexpression of the activated form of the Notch receptor (NICD), are able to attract the ISN, suggesting that they are a source of guiding signals. However, because only faint sidestep transcript expression was observed in D-AMPs and Sidestep protein was not detected, it is expected that other guiding cues may be in play. It is important to notice that to visualize the attractive potential of mis-positioned D-AMPs induced pan-mesodermal expression of NICD was observed via a GAL4/UAS system known to be thermo-sensitive. High mesodermal expression of NICD induced at 29°C leads to the loss of majority of muscles but, as is shown in this study, several muscles persist in Twi-GAL4;UAS-NICD embryos incubated at 25°C, thus allowing uncouple effects of delocalized D-AMPs from potential influence of muscles loss on ISN trajectories. However, loss of D-AMPs, observed in this study in a Poxm gain-of-function context, appears to have only a minor effect on the capacity of ISN motor axons to target dorsal muscles, which are correctly innervated by the ISN in 65% of hemisegments without D-AMP. These results highlight differential requirements of AMPs for motor axons defasciculation and navigation with L-AMPs being mandatory for L-SNa branching and D-AMPs acting as guiding cells for the ISN. However, the functional significance underlying the guidance of motor nerves by muscle stem cells remains to be determined (Lavergne, 2020).
It is also important to state that the loss of L-SNa in the sidestep mutants is not fully penetrant (observed in less than 10% of hemisegments), suggesting that sidestep is not the only player in L-SNa sub-branching. This could be due to functional redundancy between several members of Side and Beat families comprising 8 and 14 members, respectively. Expression and function of Side and Beat family members remain largely unexplored, but the fact that Sidestep labels L-AMPs and its paralog, side VI, is expressed in the DL-AMPs suggests there might be a 'Side expression code' that operates in AMPs and makes them competent to interact with navigating motor axons. In support of this hypothesis, this study found that side IV, another member of the Side family, is also expressed in AMPs with a higher transcript levels detected in L-AMPs, suggesting it could contribute to the interactions between L-AMPs and the SNa. To gain insight into AMP functions of sidestep and side IV in setting interactions with motor neurons, the selective AMP-targeting tools need to be developed to generate AMP-specific mutant rescue (Lavergne, 2020).
It has previously been shown that the nervous system is required for the establishment of the adult muscle pattern and that motor axons serve as a support for migration of AMPs during larval and pupal development. More recently, it has also been suggested that the nervous system could be involved in the selection of founder cells from the pool of AMPs. This study took advantage of a previously described genetic context (pan-muscular expression of NetB) to affect the SNa and tested impact of SNa loss on L-AMPs. In stage 16 DUF>NetB embryos, loss of the SNa observed in 84% of the hemisegments does not affect the number of L-AMPs. However, in surviving 3rd instar larvae in hemisegments lacking the SNa, number of L-AMPs is dramatically reduced. Interestingly, a specific depletion was observed of normally associated with SNa anterior L-AMPs (complete loss in 10 out of 26 hemisegments analyzed), while the posterior L-AMPs associated with the transverse nerve (TN) remained unaffected. Thus, this data provides evidence for a cross-talk between AMPs and motor axons, with the AMPs attracting navigating motor axons, which in turn are required for AMP maintenance during larval stages. The loss of anterior L-AMPs in SNa-devoid larva suggests that SNa-derived signals promote survival of associated L-AMPs, but precise underlying mechanisms remain to be elucidated (Lavergne, 2020).
Thus, in Drosophila, the dynamic interactions and close association between AMPs and the motor axon network contribute to setting ISN trajectory and are required for SNa sub-branching and proper innervation of lateral muscles, which is itself needed in larvae for the maintenance of anterior L-AMPs. In vertebrates, it has previously been described that muscle pioneers can impact motor axon pathfinding in zebrafish and more recently in mice that muscle stem cells activate and contribute to neuromuscular junction regeneration in response to denervation, and that depletion of muscle stem cells induced neuromuscular junction degeneration. This study, conducted in Drosophila, represents the first demonstration that, during development of neuromuscular system, muscle stem cells interact with motor neurons and contribute to proper muscle innervation (Lavergne, 2020).
Off-track receptor tyrosine kinase (OTK) has been shown to play an important role in the Drosophila motor axon pathfinding. The results of biochemical and genetic interactions previously suggested that OTK acts as a component of Semaphorin-1a/Plexin A (Sema-1a/PlexA) signaling during embryonic motor axon guidance and further showed that OTK binds to Wnt family members Wnt2 and Wnt4 and their common receptor Frizzled (Fz). However, the molecular mechanisms underlying the motor axon guidance function of OTK remain elusive. This study concludes that OTK mediates the forward and reverse signaling required for intersegmental nerve b (ISNb) motor axon pathfinding and it was also demonstrated that the loss of two copies of Sema-1a synergistically enhances the bypass phenotype observed in otk mutants. Furthermore, the amorphic wnt2 mutation resulted in increased premature branching phenotypes, and the loss of fz function caused a frequent inability of ISNb motor axons to defasciculate at specific choice points. Consistent with a previous study, wnt4 mutant axons were often defective in recognizing target muscles. Interestingly, the bypass phenotype of otk mutants was robustly suppressed by loss of function mutations in wnt2, wnt4, or fz. In contrast, total ISNb defects of otk were increased by the loss-of-function alleles in wnt2 and wnt4, but not fz. These findings indicate that OTK may participate in the crosstalk between the Sema-1a/PlexA and Wnt signaling pathways, thereby contributing to ISNb motor axon pathfinding and target recognition (Nguyen, 2022).
The paths axons travel to reach their targets and the subsequent synaptic connections they form are highly stereotyped. How cell surface proteins (CSPs) mediate these processes is not completely understood. The Drosophila neuromuscular junction (NMJ) is an ideal system to study how pathfinding and target specificity are accomplished, as the axon trajectories and innervation patterns are known and easily visualized. Dpr10 is a CSP required for synaptic partner choice in the neuromuscular and visual circuits and for axon pathfinding in olfactory neuron organization. This study shows that Dpr10 is also required for motor axon pathfinding. To uncover how Dpr10 mediates this process, immunoprecipitation followed by mass spectrometry were used to identify Dpr10 associated proteins. One of these, Nocte, is an unstructured, intracellular protein implicated in circadian rhythm entrainment. nocte expression in larvae was mapped; it was found to be widely expressed in neurons, muscles, and glia. Cell-specific knockdown suggests nocte is required presynaptically to mediate motor axon pathfinding. Additionally, nocte and dpr10 genetically interact to control NMJ assembly, suggesting that they function in the same molecular pathway. Overall, these data reveal novel roles for Dpr10 and its newly identified interactor, Nocte, in motor axon pathfinding and provide insight into how CSPs regulate circuit assembly (Lobb-Rabe, 2022).
N6-methyladenosine (m(6) A) regulates a variety of physiological processes through modulation of RNA metabolism. This modification is particularly enriched in the nervous system of several species, and its dysregulation has been associated with neurodevelopmental defects and neural dysfunctions. In Drosophila, loss of m(6) A alters fly behavior, albeit the underlying molecular mechanism and the role of m(6) A during nervous system development have remained elusive. This study finds that impairment of the m(6) A pathway leads to axonal overgrowth and misguidance at larval neuromuscular junctions as well as in the adult mushroom bodies. Ythdf was identified as the main m(6) A reader in the nervous system, being required to limit axonal growth. Mechanistically, this study showed that the m(6) A reader Ythdf directly interacts with Fmr1, the fly homolog of Fragile X mental retardation RNA binding protein (FMRP), to inhibit the translation of key transcripts involved in axonal growth regulation. Altogether, this study demonstrates that the m(6) A pathway controls development of the nervous system and modulates Fmr1 target transcript selection (Worpenberg, 2021).
The axon initial segment (AIS) is a compartment that serves as a molecular barrier to achieve axon-dendrite differentiation. Distribution of specific proteins during early neuronal development has been proposed to be critical for AIS construction. However, it remains unknown how these proteins are specifically targeted to the proximal axon within this limited time period. This study revealed spatiotemporal regulation in mice driven by the microtubule (MT)-based motor KIF3A/B/KAP3 (see Drosophila Klp64D) that transports TRIM46 (see Drosophila Trim9), influenced by a specific MARK2 (Drosophila homolog: Par-1) phosphorylation cascade. In the proximal part of the future axon under low MARK2 activity, the KIF3/KAP3 motor recognizes TRIM46 as cargo and transports it to the future AIS. In contrast, in the somatodendritic area under high MARK2 activity, KAP3 phosphorylated at serine 60 by MARK2 cannot bind with TRIM46 and be transported. This spatiotemporal regulation between KIF3/KAP3 and TRIM46 under specific MARK2 activity underlies the specific transport needed for axonal differentiation (Ichinose, 2019).
A neuron is a differentiated and polarized cell that consists of a cell body, several dendrites, and a long axon. To achieve the functional polarized morphology, immature neurons undergo spatiotemporal differentiation processes crucial for axon specification: outgrowth of the future axon followed by the construction of the axon initial segment (AIS), with its specific components to facilitate and maintain axonal differentiation (Ichinose, 2019).
Microtubules (MTs) are extended polymers composed of ~13 protofilaments that consist of alphaand beta-tubulin dimers whose asymmetric structure produces the polarity of MTs with a fast-growing/shrinking plus end and a less dynamic minus end. The polarity of MTs contributes to polarized intracellular trafficking as rails of motor proteins, such as kinesin super family proteins (KIFs) and dynein. KIF3, a member of the kinesin-2 family, forms a heterotrimeric complex that consists of KIF3A, KIF3B, and its cargo-binding adaptor, kinesin-associated protein 3 (KAP3) encoded by kifap3. KIF3/KAP3 is also reported to contribute to MT organization as well as cargo transport. Thus, KIF3/KAP3 has a fundamental property to produce the polarized morphology of the cells as well as the specific distributions of organelles and proteins via elaborate coordination of related molecules, including MTs (Ichinose, 2019).
Previously, the molecular interplay of cytoskeletal proteins has been proposed to be a critical intrinsic factor to generate axonal polarity in developing neurons. In developing axons, the specific proteins are transported into the future AIS and organized to construct the unique compartment, in which MTs are oriented uniformly with the plus ends toward the distal axon and the minus ends toward the cell body (plus-end out MTs). In contrast, dendritic MTs, especially in the proximal region, display a mixed-polarity orientation (mixed-polarity MTs). To date, various models and molecules have been proposed to participate in the construction of the specific MT orientation in neurons. TRIM46 belongs to the class I members of the tripartite motif (TRIM) family. It comprises an N-terminal RING finger, which is a general feature of E3 ubiquitin ligases, a B-box, a coiled-coil, a C-terminal subgroup one signature (COS) box, and a C-terminal FN3 and B30.2-like domain. The TRIM family generally consists of large protein complexes that possess ubiquitin-protein isopeptide ligase activity. In the neuron, TRIM46 is reported to have unique MT crosslinking activity localized in the most proximal part of the axon, the AIS. TRIM46 contributes to axon specification and the establishment of neuronal polarity by building closely spaced, parallel, stabilized MT fascicles in a uniformly 'plus-end out' orientation. This specific MT orientation and stability is thought to be a directional cue for the polarized trafficking of cargoes by motor proteins into the axon. However, it has been unclear how the spatiotemporal distribution of TRIM46 is produced under the elaborate coordination of specific molecules during early neuronal developmental stages. This study investigated the spatiotemporal regulation of TRIM46 accumulation at the AIS driven by the MT-based motor KIF3A/B/KAP3 under the specific MARK2 phosphorylation cascade (Ichinose, 2019).
This study reports one of the spatiotemporal mechanisms underlying AIS construction, which is a critical process for axon establishment and neuronal development. At the proximal part of the future axon, in which a low amount of MARK2 is localized, nonphosphorylated KAP3 loads TRIM46 and transports it via KIF3/KAP3 along MTs to the limited region of the future AIS. TRIM46 facilitates plus-end-out MT orientation and axon specification. In contrast, in the dendrite, in which the amount of MARK2 is highly enriched, TRIM46 does not accumulate because KAP3 phosphorylated via MARK2 cannot bind with and transport TRIM46. This study has revealed that phosphorylation via MARK2 alters the protein-protein interaction between KIF3/KAP3 and TRIM46, which directly facilitates the formation of cell polarity (Ichinose, 2019).
Intracellular transport of cargoes generally requires specific regulation for 'cargo loading' and 'cargo unloading.' As shown in this study, KIF3/KAP3 can 'load' TRIM46 as its specific cargo bound for the future AIS via the transient nonphosphorylated form of KAP3 only at approximately day in vitro (DIV) 4 (DIV4). Interestingly, this strict regulation via phosphorylation might not affect the direction and localization of KIF3 itself because both the KAP3 S60A and S60E mutants localized in axons. The regulated phosphorylation would instead affect the cargo loading activity of KIF3/KAP3. For spatiotemporal regulation, MARK2 localized in the somatodendritic region at DIV4 and later. This specific localization is also explained by previous studies showing that KIF3 initially transports the PAR-3/PAR-6/aPKC complex to the future axon, which suppresses MARK2 activity at the future axon during early developmental stages. Downregulation of MARK2 activity would facilitate the loading of TRIM46 by KAP3 and induce transport to the proximal part of the future axon to build up the AIS. This regulation of the KIF3/KAP3/TRIM46 complex would form the positive feedback loop of loading and transport of specific cargoes to further accelerate axon differentiation. However, it remains unknown how KIF3/KAP3 can 'unload' TRIM46 at the AIS. From the global localization of the KIF3/KAP3/TRIM46 complex on MTs in COS-7 cells, it is speculated that KIF3/KAP3/TRIM46 itself does not distinguish the specific structural destination cues. Instead, other regulators would be required to 'unload' TRIM46 from KIF3/KAP3 outside of the AIS to accumulate TRIM46 at only the AIS. The most likely candidate is the phosphorylation of TRIM46 by CDK5. Results from an in vitro kinase assay against TRIM46 indicated that the N-terminal region of TRIM46 containing the RING domain is phosphorylated by CDK5. S106 was identified in the RING domain as a phosphorylation site by CDK5. S106 is specific to the TRIM46 subfamily and is not conserved in the other TRIM subfamilies, such as TRIM36. Interestingly, this RING domain is reported to be necessary for TRIM46 accumulation at the AIS. In addition, CDK5 is also reported to be a key regulator of the AIS. More evidence is required to elucidate the precise unloading mechanism of TRIM46 under the CDK5 cascade (Ichinose, 2019).
Interestingly, this study found that both KIF3A and KAP3 were reduced in the kif3b+/- neurons. There should be a balance between KIF3A/B and KAP3 maintained at steady state, of which the ratio was previously reported to be 1:1:0.7, and the excess amount might be proteolyzed in the kif3b+/- neurons. Indeed, this stuy also overexpressed recombinant tagged KAP3 in neuronal cells, but only low expression of KAP3 was observed. Therefore, the strict balanced expression would explain the low coefficiency between tagged KAP3 and TRIM46 compared to that between endogenous KAP3 and TRIM46. However, tagged KAP3 was observed much more clearly in the kif3b+/- neuron than in the kif3b+/+ neuron because the expression level of KIF3 was potentially decreased to half, and there might be extra room for overexpressed KIF3 in KIF3/KAP3 balance. It might be interesting to investigate how KIF3 heterotrimers maintain their molecular balance in the future (Ichinose, 2019).
Depletion of KAP3 and KIF5 affected the distribution of AIS components. KIF5 has been reported to transport AnkG, which anchors βIV spectrin and other membrane proteins, such as Voltage-gated sodium channel (NaV) and L1-CAM, at the AIS. Because accumulated AnkG maintains axon/dendrite differentiation, KIF5 may facilitate axon differentiation by transporting AnkG. In fact, the data showing that depletion of KIF5 reduced Nav accumulation at the AIS are consistent with these previous studies. However, the detailed mechanism by which KIF5 transports AnkG to the AIS is still unknown, although it has been previously shown that KIF5 prefers stabilized MTs enriched in the AIS. More detailed mechanisms between KIF5 and AnkG as well as the regulation between KIF5 and TRIM46-stabilized MTs at the AIS should be investigated to solve this problem. However, KAP3 depletion also causes the reduction of Nav accumulation at the AIS. Moreover, it also causes Nav accumulation at the distal axon and growth cone in severe cases, which phenocopies depletion of spectrin. Considering that KIF3 transports αII spectrin, our results are consistent with previous results. Because αII spectrin is able to dimerize with either βIV spectrin at the AIS or βII spectrin at the distal axon, KIF3 may facilitate not only AIS construction but also whole axon differentiation by transporting its cytoskeletal and membrane proteins, such as axonal voltage-gated potassium channels. However, another study suggests that TRIM46 affects AIS construction by coordinating with AnkG. Depletion of AnkG reduced TRIM46 accumulation at the AIS and vice versa. The detailed mechanism between TRIM46, AnkG, KIF3, and KIF5 should be clarified to reveal the crosstalk between these molecules for the construction of the AIS. For example, it would be interesting to test whether KAP3 transports TRIM46 to the AIS in AnkG-depleted neurons. Considering that KIF5 might prefer TRIM46-stabilized MTs, there would be positive feedback assembly of the AIS by cooperative transport between KIF5 and KIF3. This molecular coordination would set up a selective barrier for the polarized trafficking of vesicles and organelles into the axon; axon/dendrite differentiation is further accelerated via previously reported mechanisms. Another major MT motor, KIF1A, does not seem to regulate axonal differentiation because the localization of Nav at the AIS was not affected by KIF1A depletion. However, the length of the AIS visualized with Nav immunostaining was slightly increased. Increased AIS length was also observed in auditory-deprived chick neurons. Considering that KIF1A transports synaptic vesicle precursors, it is possible that reduced presynaptic activity by KIF1A depletion increased the AIS length (Ichinose, 2019).
As shown in this study, the spatiotemporal molecular interplay of KIF3/KAP3/TRIM46 under MARK2 phosphorylation facilitates axon specification. AIS construction is important not only for selective transport but also for initiation of action potentials. Thus, manipulating neuronal excitability in this region would be a potential target for the treatment of epilepsy and psychiatric disorders. To reveal the relation between the regulation of AIS construction and neuronal diseases, further precise analyses, including both electrophysiological approaches in vivo and structural in vitro reconstruction studies, will contribute to a deeper understanding (Ichinose, 2019).
Spatiotemporal mechanisms generating neural diversity are fundamental for understanding neural processes. This study investigated how neural diversity arises from neurons coming from identical progenitors. In the dorsal thorax of Drosophila, rows of mechanosensory organs originate from the division of sensory organ progenitor (SOPs). In each row of the notum, an anteromedial located central SOP divides first, then neighbouring SOPs divide, and so on. This centrifugal wave of mitoses depends on cell-cell inhibitory interactions mediated by SOP cytoplasmic protrusions and Scabrous, a secreted protein interacting with the Delta/Notch complex. Furthermore, when this mitotic wave was reduced, axonal growth was more synchronous, axonal terminals had a complex branching pattern and fly behaviour was impaired. The temporal order of progenitor divisions influences the birth order of sensory neurons, axon branching and impact on grooming behaviour. These data support the idea that developmental timing controls axon wiring neural diversity (Lacoste, 2022).
To study how functional neuronal diversity can be generated from a homogenous set of neural precursors, advantage was taken of the invariant way in which sensory organs are located on the dorsal epithelium of Drosophila. This spatial configuration greatly facilitated the study of the relative timing of SOP division and the identification of a distinct temporal wave of SOP mitosis. Asynchrony in mitotic reactivation timing has been described in Drosophila larva neuroblasts. This differential timing is related to two cell cycle arrests: one population of neuroblasts is arrested in G2 while another population is arrested in G0. G2-arrested neuroblasts resume mitosis earlier than those in G0-arrest. As in this system, it has been proposed that this particular order of division ensures that neurons form appropriate functional wiring. It is relevant that other temporal processes controlling the wiring of peripheral receptors with the central nervous system have been described in the Drosophila eye, another highly organised structure. It is conceivable that these temporal patterning mechanisms of neurogenesis, to date identified only in organised tissues, could be more widespread (Lacoste, 2022).
A core aspect of this work was to link cellular level of complexity (timing of SOP division) with uppermost level (behaviour). In this context, evidences are presented showing that the cleaning reflex was impaired when the SOP mitotic wave was disrupted. The cleaning reflex has been traditionally analysed after stimulation of macrochaetes rather than microchaetes as in the present work. Macro- and microchaetes have different patterns of terminal axon arborisation (Usui-Ishihara and Simpson, 2005). As such, it is remarkable that this fly behaviour was significantly affected by altering the timing of microchaete precursor division in the dorsal thorax. This study showed that the SOP mitotic wave leads to a progressive neurogenesis along each row of microchaetes. This, in turn, would likely induce a particular pattern of microchæte axon arrival in the thoracic ganglion required for the proper organisation of the neuropila in the central nervous system. Although this study has documented this progressive axonogenesis, the strict pattern of axon arrival into the ventral ganglion is not known. It would depend on the order of birth of neurons, and on the geometry of axon projections that fasciculate to form the dorsal mesothoracic nerves in the ganglion. In any case, this study shows that, when sca function was specifically downregulated during the SOP mitotic wave, axonogenesis occurs almost simultaneously in each row of microchaetes. This certainly impairs the pattern of axon arrival into the ganglion leading to ectopic axon branching and changes in fly behaviour. It would be interesting to know whether these impairments are specifically due to neurogenesis occurring simultaneously. To test this, it is necessary to find a way to induce different patterns of SOP mitotic entry, for instance, a centripetal wave or a random order. If the observed effect is specifically due to the simultaneity, normal behaviour would be expected to be associated with other patterns of SOP division (Lacoste, 2022).
We observed that the first SOP to divide (SOP0) was always located in the anteromedial region of each row. This may reflect the existence of a pre-pattern that causes SOPs located in that region to start dividing earlier than the others. Although the anteromedial region corresponds approximately to the posterior limit of expression of the transcription factor BarH1, no factors specifically expressed in this region have yet been identified. Alternatively, as the location of SOP0 is modified when Sca function was impaired, an interesting possibility is that SOP0 is selected by an emergent process related to cell-cell interaction in the epithelium, rather than by a passive pre-pattern that organises the first events in the notum (Lacoste, 2022).
This study presents evidence indicating that the secreted glycoprotein Scabrous, which is known to interact with the N-pathway to promote neural patterning, controls the kinetics of SOP mitosis in the notum. In proneural clusters, cells that express high levels of Dl and Sca become SOPs, while surrounding epithelial cells activate the N-pathway to prevent acquisition of a neural fate. In eye and notum systems, Sca modulates N-activity at a long range. Indeed, during eye development, sca is expressed in intermediate clusters in the morphogenic furrow and transported posteriorly in vesicles through cellular protrusions to negatively control ommatidial cluster rotation. Similarly, in the notum, SOP protrusions extend beyond several adjacent epithelial cells in which Dl and Scabrous are detected. The current data show that shorter protrusions (obtained after rac1N17 overexpression conditions) as well as loss of function of Dl or sca make the mitotic wave more synchronous. Since, no reduction of the global level of sca expression associated with the wave progression was observed, it is plausible that Sca, required to maintain SOPs in G2 arrest, is delivered focally through protrusions that are difficult to follow with in vivo analysis. Although this possibility is favored, it cannot be formally ruled out that Rac1N17 overexpression affects Sca secretion per se without affects sca expression (Lacoste, 2022).
As in neuroblasts, G2 arrest in SOP cells is due to the downregulation of the promitotic factor Cdc25/String. Thus, overexpression of string in SOPs induces a premature entry into mitosis, while overexpression of negative regulators, like Wee1, maintain these cells in arrest. Possibly Sca negatively regulates string expression, perhaps through the N-pathway that it is known to control the level of String. Alternatively, it has been recently shown that the insulin-pathway also regulates String level. Moreover, in muscle precursors, cell proliferation is induced by the insulin-mediated activation of the N-pathway. These observations raise the interesting possibility that, in this system, insulin activates the N-pathway and Sca modulates this activation. Further investigations will be required in order to identify the link between Scabrous, the N/Dl- and insulin-pathways in the resumption of mitosis in SOPs (Lacoste, 2022).
During nervous system development, the complex patterns of neuronal wiring are achieved through the interaction between neuronal cell surface receptors and their chemoattractive or repulsive ligands present in the environment. An essential condition for proper axon guidance is the competence of neurons to respond to these environmental clues. It is generally agreed that neuron competence depends on the specific expression of transcriptional factors regulating their identity. This study shows that the timing of neuron formation is also a factor controlling their terminal morphology. It is proposed that the SOP mitotic wave induces a particular pattern of arrival of microchæte axons in the thoracic ganglion. This pattern establishes a specific framework of guidance cues on which circuits will be built and ultimately influencing an organism's behaviour. These findings support the idea that, in addition to genetic factors, neurogenic timing is a parameter of development in the mechanisms controlling neural branching (Lacoste, 2022).
The exocyst complex is an important regulator of intracellular trafficking and tethers secretory vesicles to the plasma membrane. Understanding of its role in neuron outgrowth remains incomplete, and previous studies have come to different conclusions about its importance for axon and dendrite growth, particularly in vivo. To investigate exocyst function in vivo Drosophila sensory neurons were used as a model system. To bypass early developmental requirements in other cell types, neuron-specific RNAi was used to target seven exocyst subunits. Initial neuronal development proceeded normally in these backgrounds, however, this was considered to be due to residual exocyst function. To probe neuronal growth capacity at later times after RNAi initiation, laser microsurgery was used to remove axons or dendrites and prompt regrowth. Exocyst subunit RNAi reduced axon regeneration, although new axons could be specified. In control neurons, a vesicle trafficking marker often concentrated in the new axon, but this pattern was disrupted in Sec6 RNAi neurons. Dendrite regeneration was also severely reduced by exocyst RNAi, even though the trafficking marker did not accumulate in a strongly polarized manner during normal dendrite regeneration. The requirement for the exocyst was not limited to injury contexts as exocyst subunit RNAi eliminated dendrite regrowth after developmental pruning. It is concluded that the exocyst is required for injury-induced and developmental neurite outgrowth, but that residual protein function can easily mask this requirement (Swope, 2022).
Axonal growth is mediated by coordinated changes of the actin and microtubule (MT) cytoskeleton. Ample evidence suggests that members of the formin protein family are involved in the coordination of these cytoskeletal rearrangements, but the molecular mechanisms of the formin-dependent actin-microtubule crosstalk remains largely elusive. Of the six Drosophila formins, DAAM was shown to play a pivotal role during axonal growth in all stages of nervous system development, while FRL was implicated in axonal development in the adult brain. This study aimed to investigate the potentially redundant function of these two formins, and attempts were made to clarify which molecular activities are important for axonal growth. A combination of genetic analyses, cellular assays and biochemical approaches was used to demonstrate that the actin-processing activity of DAAM is indispensable for axonal growth in every developmental condition. In addition, a novel MT-binding motif was identified within the FH2 domain of DAAM, which is required for proper growth and guidance of the mushroom body axons, while being dispensable during embryonic axon development. Together, these data suggest that DAAM is the predominant formin during axonal growth in Drosophila, and highlight the contribution of multiple formin-mediated mechanisms in cytoskeleton coordination during axonal growth (Foldi, 2022).
A combination of intermittent active movement of transient aggregates and a paused state that intervenes between periods of active transport has been proposed to underlie the slow, directed transport of soluble proteins in axons. A component of passive diffusion in the axoplasm may also contribute to slow axonal transport, although quantitative estimates of the relative contributions of diffusive and active movement in the slow transport of a soluble protein, and in particular how they might vary across developmental stages, are lacking. This work proposes and studies a model for slow axonal transport, addressing data from bleach recovery measurements on a small, soluble, protein, choline acetyltransferase, in thin axons of the lateral chordotonal (lch5) sensory neurons of Drosophila. Choline acetyltransferase is mainly present in soluble form in the axon and catalyzes the acetylation of choline at the synapse. It does not form particulate structures in axons and moves at rates characteristic of slow component b (≈ 1-10 mm/day or 0.01-0.1 &mi;m/s). Using this model, which incorporates active transport with paused and/or diffusive states, bleach recovery, transport rates, and cargo trajectories obtained through kymographs were predicted, comparing these with experimental observations at different developmental stages. Changes were shown in the diffusive fraction of cargo during these developmental stages dominate bleach recovery, and a combination of active motion with a paused state alone could reproduce the data. Predictions of the model were compared with results from photoactivation experiments. The importance of the diffusive state in reproducing the bleach recovery signal in the slow axonal transport of small soluble proteins is the central result of this study (Maiya, 2023).
Axon guidance cues direct the growth and steering of neuronal growth cones, thus guiding the axons to their targets during development. Nonetheless, after axons have reached their targets and established functional circuits, many mature neurons continue to express these developmental cues. The role of axon guidance cues in the adult nervous system has not been fully elucidated. Using the expression pattern data available on FlyBase, this study found that more than 96% of the guidance genes that are expressed in the Drosophila melanogaster embryo continue to be expressed in adults. The GeneSwitch and TARGET systems were used to spatiotemporally knockdown the expression of these guidance genes selectively in the adult neurons, once the development was completed. An RNA interference (RNAi) screen was performed against 44 guidance genes in the adult Drosophila nervous system, and 14 genes were identified that are required for adult survival and normal motility. Additionally, it was shown that adult expression of Semaphorins and Plexins in motor neurons is necessary for neuronal survival, indicating that guidance genes have critical functions in the mature nervous system (Vaikakkara Chithran, 2023).
Notch-dependent binary fate choice between sister neurons is one of the mechanisms to generate neural diversity. How these upstream neural fate specification programs regulate downstream effector genes to control axon targeting and neuropil assembly remains less well understood. This tudy reports that Notch-dependent binary fate choice in Drosophila medulla neurons is required to regulate the Netrin axon guidance pathway, which controls targeting of transmedullary (Tm) neurons to lobula. In medulla neurons of Notch-on hemilineage composed of mostly lobula-targeting neurons, Notch signaling is required to activate the expression of Netrin-B and repress the expression of its repulsive receptor Unc-5. Turning off Unc-5 is necessary for Tm neurons to target lobula. Furthermore, Netrin-B provided by Notch-on medulla neurons is required for correct targeting of Tm axons from later-generated medulla columns. Thus, the coordinate regulation of Netrin pathway components by Notch signaling ensures correct targeting of Tm axons and contributes to the neuropil assembly (Zhang, 2023).
During nervous system development, neural diversity is generated through integration of temporal patterning, spatial patterning, and Notch-dependent binary fate choice between sister neurons. How these upstream neural specification programs are translated into highly organized neuropils is not well understood. In this work, using the Drosophila visual system as an example, it was demonstrated that Notch signaling regulates the expression of Netrin pathway components in Notch-on hemilineage neurons, and that the Netrin pathway controls targeting of visual projection neurons (Tm) to the higher order neuropil (lobula). Notch signaling is required to repress Unc-5 expression in Tm neurons. The absence of Unc-5 is essential for Tm neurons to target lobula, and also required for Tm neurons to further enrich NetB in lobula using the Fra receptor. Furthermore, Notch signaling is required to activate NetB expression in Tm neurons, and medulla-derived NetB has an essential role in promoting targeting of Tm axons from later-born medulla columns to lobula through the IOC. Yhe regulation of NetB distribution and the regulation of Netrin pathway components by Notch signaling and possibly other factors is discussed (Zhang, 2023).
Previous studies found that, in other neuropils, Fra captures NetB to enrich Netrin pathway components locally. This study also found that Fra is colocalized with NetB in lobula neuropil layers. Without Fra expressed in medulla projection neurons, NetB enrichment at lobula is greatly diminished. However, since Fra also guides the targeting of Tm neurons to lobula and Tm neurons are also part of the Netrin source, it is possible that both mechanisms contribute to the diminished NetB in the lobula (Zhang, 2023).
Although Fra is required for NetB enrichment, not all Fra is colocalized with NetB in the optic lobe. Furthermore, Unc-5 may be a negative regulator of NetB enrichment. In unc-5 mutants, NetB spreads to lobula plate and medulla. Moreover, overexpression of Unc-5 results in disrupted NetB distribution. However, how Unc-5 regulates NetB distribution requires future investigation. Unc-5 may affect NetB distribution directly or indirectly by affecting the guidance of specific neurons, which affect NetB distribution (Zhang, 2023).
Since mis-expressing Unc-5 in Tm neurons results in their repulsion to the lobula plate, Unc-5 expression should be strictly inhibited in Tm neurons during axon targeting. Unc-5 expression was found to be activated ectopically without Notch signaling, likely in most Tm neurons. However, the regulation of Unc-5 expression in medulla neurons is far more complex and may involve multiple factors/mechanisms. First, besides Tm neurons, Notch-on hemilineage also includes Mi1 neurons, which terminate at the proximal medulla neuropil, and they do keep expressing Unc-5. Therefore, Notch signaling does not inhibit Unc-5 expression in all Notch-on neurons, and in these exceptions, like Mi1 neurons, other factors, such as transcriptional/translational feedback (TTF) or neuronal-specific transcription factors, might override the inhibition of Unc-5 expression by Notch signaling. Consistent with this hypothesis, loss of Hth or Bsh transforms Mi1 into a Tm-type neuron. Second, there should be other factors or mechanisms that repress Unc-5 expression in medulla neurons. A middle layer of neurons (likely born in and around the Ey temporal stage) were observed that turn off Unc-5 regardless of their Notch status. Although most Tm and TmY neurons belong to the Notch-on hemilineage, there are a few exceptions; for example, scRNA-seq data showed that Tm5c, Tm29, and a few TmY neuron types do not express Ap. Among them, Tm29, TmY5a, and TmY14 are shown to express Knot (Kn), and are thus generated from the Notch-off hemilineage in the Ey stage (corresponding to the middle-layer gap in unc-5 expression). Thus, these neurons likely turn off Unc-5 by a different mechanism independent of Notch signaling. It will be interesting to examine whether the TTF(s) or neuronal transcription factors such as Kn are involved. Third, while Unc-5 is expressed in most young immature neurons, it is turned off later in Notch-on neurons, suggesting that a later-acting factor that is specifically activated in the Notch-on hemilineage is required to turn off Unc-5 expression (Zhang, 2023).
In addition to repression of Unc-5, this study also showed that Notch signaling is required to activate NetB expression in Notch-on medulla neurons. Thus, the results suggest that Notch-dependent binary fate choice ensures that lobula-targeting neurons will turn on NetB expression and provide more NetB to the lobula NetB pool. Tm axons from later-born posterior medulla columns may require a higher concentration of NetB to target lobula, because of the longer distance they need to migrate. Consistently, when medulla-derived NetB was knocked down using SoxN-Gal4, Tm axons from posterior columns showed disrupted IOC formation (Zhang, 2023).
Taken together, these results suggest that, in the Notch-on hemilineage neurons, Notch signaling is required to repress the repulsive receptor Unc-5 and also to activate the expression of NetB, which ensures that projection neurons targeting lobula can further enrich NetB with their Fra receptors as well as secrete more NetB. This positive feedback mechanism is required for the correct formation of interommatidial cells (IOC) through which visual project neurons connect to their target neuropil. Thus, Notch-dependent binary fate choice in medulla neurons coordinately regulates the expression of an axon guidance cue and its receptors, which is required for the correct assembly of neuropils (Zhang, 2023).
This study demonstrated that Notch signaling is required to regulate Netrin pathway components. However, it is not known whether this regulation is direct or indirect. In addition, the regulation of Netrin pathway components is complex, and should involve integration of Notch signaling with other factors such as temporal factors and spatial factors (Zhang, 2023).
Ena/VASP proteins are processive actin polymerases that are required throughout animal phylogeny for many morphogenetic processes, including axon growth and guidance. This study used in vivo live imaging of morphology and actin distribution to determine the role of Ena in promoting the growth of the TSM1 axon of the Drosophila wing. Altering Ena activity causes stalling and misrouting of TSM1. These data show that Ena has a substantial impact on filopodial morphology in this growth cone but exerts only modest effects on actin distribution. This is in contrast to the main regulator of Ena, Abl tyrosine kinase, which was shown previously to have profound effects on actin and only mild effects on TSM1 growth cone morphology. These data are interpreted as suggesting that the primary role of Ena in this axon may be to link actin to the morphogenetic processes of the plasma membrane, rather than to regulate actin organization itself. These data also suggest that a key role of Ena, acting downstream of Abl, may be to maintain consistent organization and reliable evolution of growth cone structure, even as Abl activity varies in response to guidance cues in the environment (Fang, 2023).
Most invertebrate axons and small caliber axons in mammalian peripheral nerves are unmyelinated but still ensheathed by glia. This study used Drosophila wrapping glia to study the development and function of non-myelinating axon ensheathment, which is poorly understood. Selective ablation of these glia from peripheral nerves severely impaired larval locomotor behavior. In an in vivo RNAi screen to identify glial genes required for axon ensheathment, the conserved receptor tyrosine kinase Discoidin domain receptor (Ddr) was identified. In larval peripheral nerves, loss of Ddr resulted in severely reduced ensheathment of axons and reduced axon caliber, and a strong dominant genetic interaction was found between Ddr and the type XV/XVIII collagen Multiplexin (Mp), suggesting Ddr functions as a collagen receptor to drive axon wrapping. In adult nerves, loss of Ddr decreased long-term survival of sensory neurons and significantly reduced axon caliber without overtly affecting ensheathment. These data establish essential roles for non-myelinating glia in nerve development, maintenance, and function, and identify Ddr as a key regulator of axon-glia interactions during ensheathment and establishment of axon caliber (Corty, 2022).
Non-myelinating ensheathment of axons is a conserved but understudied feature of the PNS. Although this type of multi-axonal ensheathment has been less studied compared with myelination, a growing body of evidence indicates it is important for the health and function of neurons and axons in the periphery. For example, Schwann cell-specific loss of the transmembrane receptor LDL receptor related protein-1 (LRP1) causes both thin myelin and abnormal Remak bundle structure. These conditional knockout animals also showed a lowered pain threshold, suggesting that the physiology of nociceptor neurons is impaired when Remak ensheathment is disrupted. Disrupting metabolism in Schwann cells causes progressive axon loss, with small unmyelinated fibers dying first, before myelinated fibers begin to show signs of degeneration. In the fly, disruption of axonal wrapping leads to uncoordinated behavioral responses that hint at aberrant ephaptic coupling between neighboring axons in nerves when not properly separated (Kottmeier, 2020). Such coupling could cause the inappropriate activation of sensory or nociceptive neurons underlying peripheral neuropathies. Previous studies lab have shown that wrapping glia are required to clear neuronal debris after nerve injury and mediate injury signaling between injured and intact 'bystander' neurons, which might be important for functional recovery after nerve trauma. These and other findings suggest that Remak-type ensheathment and axon-glia signaling of unmyelinated fibers play a variety of underappreciated roles in peripheral nerve physiology that contribute to the pathophysiology of a number of PNS disorders, including debilitating peripheral neuropathies and responses to nerve injury (Corty, 2022).
To gain insight into non-myelinating ensheathment, the Drosophila peripheral nerves were used to identify a molecular pathway important for the development and function multi-axonal ensheathment. A new Split-Gal4 intersectional driver was generated to target wrapping glia more specifically for functional and behavioral studies in order to improve understanding of whether and how wrapping glia support axon health, physiology and, ultimately, circuit function. Finally, this study uncovered roles for glia in mediating long-term neuronal survival and driving increased axon caliber that are separable from overt effects on wrapping, demonstrating that non-myelinating ensheathing glia perform crucial, previously unappreciated, roles in nervous system development, maintenance and function (Corty, 2022).
A main advantage of Drosophila is the ability to conduct large-scale in vivo screens. Use was made of available UAS-RNAi libraries to carry out a broad screen for regulators of axonal ensheathment in intact nerves. This morphological screen was sensitive enough to identify genes previously implicated in wrapping glia development, including vn, LanB1 and mys, validating the approach. Moreover, in the case of Ddr, it was possible to identify an important regulator of ensheathment that a simple behavioral or lethality screen would have missed in light of follow-up behavioral testing. Knockdown of Ddr in wrapping glia resulted in reduced glial membrane coverage in nerve cross-sections by fluorescence microscopy. Similar phenotypes were observed in Ddr loss-of-function animals and could be rescued by resupplying Ddr specifically in wrapping glia, confirming the specificity of the RNAi results. TEM clearly showed that reduced glial membrane coverage at the light level corresponds to decreased axon wrapping (Corty, 2022).
Although neither of the vertebrate homologs, Ddr1 and Ddr2, has been explicitly implicated in glial development, several lines of evidence suggests that Ddr1 may have a conserved role in vertebrate glial development or function. Ddr1 is highly expressed in the mouse oligodendrocyte lineage starting from when the cells begin to associate with axons, is upregulated in newly formed oligodendrocytes after cuprizone treatment, and is expressed in both myelinating and Remak Schwann cells. Moreover, DDR1 is expressed in human oligodendrocytes and myelin, and variants in the human gene have been correlated with abnormal white matter and schizophrenia (Corty, 2022).
Vertebrate Ddr1 and Ddr2 are potently activated by collagens in vitro, prompting an investigation of whether collagens were involved with Ddr function in fly nerves. Knockdown of the Drosophila collagen Mp specifically in wrapping glia but not in neurons was found to disrupted ensheathment. Together with the established roles for vertebrate Ddr1 and Ddr2 as collagen receptors, the strong genetic interaction observed between Ddr and Mp is consistent with a model in which Mp acts as a collagen ligand for Ddr during axonal ensheathment. Although the Mp-GFP protein trap shows diffuse Mp expression throughout the nerve, it remains unclear precisely which cell type(s) within the nerve are producing it. Previous reports indicate that Mp can be expressed in the outer peripheral glia layers, so they may provide some Mp to the wrapping glia. However, the strong ensheathment defect seen when Mp is knocked down exclusively in wrapping glia indicates that wrapping glia themselves are likely to be the primary, relevant source of the Mp required for their own morphogenesis. Schwann cells similarly rely on components of their own basal lamina to regulate their development. For example, laminin-211 serves as a ligand for GPR126 to promote myelination. Mp is the sole Drosophila homolog of collagen types XV/XVIII, containing a central helical collagen region with a cleavable N-terminal thrombospondin-like domain and C-terminal endostatin-like domain. Collagen 15a1 and 18a1 are expressed in mouse peripheral nerves and Col15a1 mutants have radial-sorting defects, suggesting that the role of Mp in promoting axon wrapping is likely conserved. In fact, Mp appears to play multiple roles in nerve biology. For example, Mp secreted by the outer glia layers acts via its cleaved endostatin domain to modulate homeostatic plasticity at motor neuron synapses. How Ddr activation within wrapping glia ultimately drives axon wrapping still remains to be determined, but Ddr joins two other receptor tyrosine kinases - EGFR and FGFR - as important and conserved regulators of axon ensheathment. As a non-canonical collagen receptor, Ddr may also interact with other collagen receptors, such as integrins (known to play roles in wrapping glia development), to sense and remodel the extracellular matrix and permit extension of glia processes between axons, similar to its roles in promoting tumor metastasis (Corty, 2022).
The nrv2-Gal4 driver has been the standard method to genetically target wrapping glia for morphological studies, but it is imperfect for manipulation of wrapping glia in ablation or behavioral assays owing to its expression in several subtypes of CNS glia. This study generated a new Split-Gal4 intersectional driver that drives exclusively in wrapping glia. This allowed performing of precise ablation of wrapping glia that led to severely impaired larval locomotion, indicating that the wrapping glia are essential for basic crawling circuit function. This phenotype was particularly striking in light of that fact that no clear crawling defect was observed in Ddr mutant larvae, even though wrapping was severely impaired. It may be possible that non-contact-mediated mechanisms, such as one or more secreted factors, constitute the essential contribution of wrapping glia to axon health and physiology. Alternatively, perhaps even a small amount of direct glia-axon contact may be sufficient to support neuron health and axon function. This would be consistent with the lack of overt behavioral defects in newly hatched first instar larvae, which have poor wrapping compared with later stages, and even in wild-type third instar larvae, in which not every axon is individually wrapped. It is also consistent with our findings that many nerves in WG-ablated larvae seem to be missing axons, whereas this was not observed in Ddr mutant nerves. These results are also similar to what has been recently reported using a different approach to ablate wrapping glia, where only minor behavioral defects were observed upon FGFR signaling disruption but profound crawling defects were seen upon ablation. As with all ablation studies, it is not possible to strictly rule out unexpected negative side effects of the ablation itself; however, using a genetic approach should limit collateral damage (compared with laser or toxin approaches). Together, these data support the conclusion that even limited wrapping or simply some degree of glia-axon contact is sufficient to support axon survival and nerve function compared with no glia at all at least for the first ~5 days of larval life (Corty, 2022).
Previous studies of oligodendrocytes and Schwann cells have found that impairing glial function can result in seemingly normal wrapping and circuit function in young animals, with deficits only appearing when the system is stressed or aged. Studying wrapping in adult Drosophila allows for aging and maintenance studies that the short larval period precludes. Adult peripheral nerves are encased in a transparent but hard cuticle that allows for live imaging but makes fixation challenging. Because of the resolution limits of light microscopy, a reliable method was developed to study their ultrastructure using TEM. Ensheathment in the adult wing nerve was found to differ from that of the larva, as all axons appear to be separated by glial membranes. Surprisingly, wrapping was not obviously impaired in adult nerves of Ddr knockdown or mutant animals. One difference between larval and adult wrapping glia is the territory size of each cell. In larvae, one wrapping glia cell covers the majority of the nerve from the VNC to the muscle field. This wrapping glial cell must therefore undergo tremendous growth to keep up with nerve elongation as the animal grows, as well as radial growth to ensheathe axons. A single cell can end up covering from ~750 μm to 2.5 mm of nerve length, depending on the segment, whereas in the wing there are ~13 wrapping glia along the region of the L1 nerve that was analyzed; This is is ~400 μm long. In larval wrapping glia, there are three receptor tyrosine kinases (EGFR, the FGFR Heartless, and now Ddr) that are each required for normal ensheathment, and thus cannot fully compensate for one another. It is hypothesized that in the larva the cell is pushed to its growth limits and any perturbation in pro-wrapping signaling has a strong effect on morphology, whereas in the adult nerve the system is robust and redundant enough to withstand perturbations of single genes. Future studies of double and triple mutants may be able to test this hypothesis (Corty, 2022).
Loss of Ddr led to an increase in spontaneous neurodegeneration in the nerve as animals naturally aged. Such an uncoupling of neuron health from overt effects on myelination has been demonstrated previously. For example, Cnp1 (Cnp) mutant mice show severe age-dependent neurodegeneration, although they have grossly normal myelin with only subtle changes in myelin ultrastructure. Loss of the proteolipid PLP results in axon degeneration despite having largely normal myelin. It was found that the number of VGlut+ neurons was reduced in aged wings of Ddr knockdown animals, indicating that wrapping glial Ddr is important for long-term neuronal survival. When Ddr whole animal mutants were analyzed by TEM a small but significant reduction was found in axon profile number, which should correspond to the number of surviving neurons. Together with the increased variability observed, this suggests that absence of Ddr signaling increases the susceptibility of subpopulations of neurons to insult or injury that may underlie age-related degeneration (Corty, 2022).
Myelination can directly affect the structure and function of the axons they wrap, including controlling caliber. In general, myelination increases caliber. For example, dysmyelinated Trembler mice have reduced axon calibers compared with controls, and in the PNS caliber along a single axon can vary with reduced caliber at points without direct myelin contact, such as nodes of Ranvier. Axon caliber is an important determinant of conduction velocity but varies widely between neuronal subtypes, so achieving and maintaining appropriate caliber is crucial for proper circuit function. How non-myelinating ensheathment impacts axon caliber is not understood. This study found glial Ddr promotes increased axon caliber. This study focused on the distal twin sensilla of the margin (dTSM) neuron, so it was possible to directly compare the caliber of an identifiable axon between conditions. The reductions in caliber were similar between Ddr mutants and glial-specific DdrRNAi, supporting a non-cell-autonomous role for glial Ddr in regulating axon caliber. The effect is considerable: nearly a 50% reduction in axon caliber at 5 dpe. We hypothesize that by this time point, wild-type dTSM axons have reached their mature caliber, as it is comparable between 5 dpe and 28 dpe in comparable genetic backgrounds. In Ddr mutants, however, we observe that the relative size compared with controls changes over time, suggesting that in Ddr mutants (or knockdowns) the axon continues to increase its caliber, perhaps in an effort to achieve the optimal size, although the axons still remain ~25% smaller than wild-type axons at 28 dpe (Corty, 2022).
Two proteins, MAG, which acts to increase the caliber of myelinated axons, and CMTM6, which restricts the caliber of myelinated and unmyelinated axons, are the only proteins reported to non-cell-autonomously affect the caliber of vertebrate axons, and both do so without overtly affecting myelin. In the fly, it has been shown that a shift in the average size of axons in larval nerves when wrapping glia are absent or severely disrupted, supporting a general role for wrapping glia in promoting axon size. In the adult, this study showed that Ddr is still required for increased axon caliber even when wrapping appears intact. The exact molecular mechanism by which Ddr may promote increased caliber size remains unclear as the control of axon caliber, generally, is not well understood. Genes involved in the general regulation of cell size have been implicated as cell-autonomous determinants. For example, in the fly, S6 kinase signaling is a positive regulator of motor neuron size, including axon caliber. In mammalian axons, the phosphorylation state of neurofilaments and microtubules determines their spacing to determine caliber. Determining how glial Ddr activity ultimately influences the axonal cytoskeleton is an important next step. A 25-50% reduction in caliber would be predicted to impact conduction velocity along the dTSM axon. Given that campaniform sensilla provide essential rapid sensory feedback to fine-tune movement, it will be of interest to test conduction velocity and flight behavior in Ddr mutant animals to see how the proprioceptive circuit might be affected (Corty, 2022).
Taken together, these studies identify Ddr as an important regulator of wrapping glia development and function in the fly, with distinct roles in larval and adult wrapping glia. Ddr is essential for the normal morphological development of axon wrapping in the larvae, and also mediates important axon-glia communication that controls axon caliber growth and affects neuronal health and survival. Given its expression pattern in vertebrate oligodendrocytes and Schwann cells, it seems likely that these essential functions are conserved in vertebrates. Further study into how Ddr functions in both fly and vertebrate glia promises to increase understanding of axon ensheathment in health and disease (Corty, 2022).
Serotonergic neurons produce extensively branched axons that fill most of the central nervous system, where they modulate a wide variety of behaviors. Many behavioral disorders have been correlated with defective serotonergic axon morphologies. Proper behavioral output therefore depends on the precise outgrowth and targeting of serotonergic axons during development. To direct outgrowth, serotonergic neurons utilize serotonin as a signaling molecule prior to it assuming its neurotransmitter role. This process, termed serotonin autoregulation, regulates axon outgrowth, branching, and varicosity development of serotonergic neurons. However, the receptor that mediates serotonin autoregulation is unknown. This study asked if serotonin receptor 5-HT1A plays a role in serotonergic axon outgrowth and branching. Using cultured Drosophila serotonergic neurons, this study found that exogenous serotonin reduced axon length and branching only in those expressing 5-HT1A. Pharmacological activation of 5-HT1A led to reduced axon length and branching, whereas the disruption of 5-HT1A rescued outgrowth in the presence of exogenous serotonin. Altogether this suggests that 5-HT1A is a serotonin autoreceptor in a subpopulation of serotonergic neurons and initiates signaling pathways that regulate axon outgrowth and branching during Drosophila development (Long, 2023).
Johnston's organ (Jo) acts as an antennal wind-sensitive and/or auditory organ across a spectrum of insect species and its axons universally project to the brain. In the locust, this pathway is already present at mid-embryogenesis but the process of fasciculation involved in its construction has not been investigated. Terminal projections into the fine neuropilar organization of the brain also remain unresolved, information essential not only for understanding the neural circuitry mediating Jo-mediated behavior but also for providing comparative data offering insights into its evolution. In this study, neuron-specific, axon-specific, and epithelial domain labels were employed to show that the pathway to the brain of the locust is built in a stepwise manner during early embryogenesis as processes from Jo cell clusters in the pedicel fasciculate first with one another, and then with the two tracts constituting the pioneer axon scaffold of the antenna. A comparison of fasciculation patterns confirms that projections from cell clusters of Jo stereotypically associate with only one axon tract according to their location in the pedicellar epithelium, consistent with a topographic plan. At the molecular level, all neuronal elements of the Jo pathway to the brain express the lipocalin Lazarillo, a cell surface epitope that regulates axogenesis in the primary axon scaffold itself, and putatively during fasciculation of the Jo projections to the brain. Central projections from Jo first contact the primary axon scaffold of the deutocerebral brain at mid-embryogenesis, and in the adult traverse mechanosensory/motor neuropils similar to those in Drosophila. These axons then terminate among protocerebral commissures containing premotor interneurons known to regulate flight behavior (Boyan, 2023).
Drosophila Robo3 is an axon guidance receptor that regulates longitudinal axon tract formation in the embryonic ventral nerve cord. Robo3 is thought to guide longitudinal axons by signaling repulsion in response to Slit. To test this, the robo3 locus was modified to express a version of the receptor lacking its cytoplasmic domain (Robo3δC). Lngitudinal axon guidance was reduced, but not eliminated, in embryos expressing Robo3ΔC. These results show that Robo3's cytodomain is partially dispensable for its axon guidance activity and suggest that it may guide axons via a mechanism other than direct transduction of Slit-dependent signaling (Agcaoili, 2024).
To examine the functional importance of the Robo3 cytodomain, CRISPR was used to engineer the robo3 locus to express a version of Robo3 lacking its cytodomain (Robo3δC). Ywo guide RNAs (gRNAs) along with a homologous donor plasmid containing 1 kb upstream and downstream homology regions were used to replace exons 2-12 of robo3 with an HA-tagged robo3δC cDNA, creating the robo3δC allele. Previously studies used a similar strategy to replace robo3 with cDNAs encoding full-length wild type robo3, its Drosophila paralogs robo1 and robo2, its ortholog from Tribolium castaneum, TcRobo2/3 and engineered robo3 variants in which the Ig1 domain was deleted or otherwise modified. These previous studies demonstrate that replacing exons 2-12 and all of the intervening introns does not impair the expression or function of robo3 in the embryonic ventral nerve cord, nor does the addition of an N-terminal 4xHA tag interfere with Slit binding or longitudinal axon guidance by Robo3 (Agcaoili, 2024).
First, the effect was examined of deleting the cytodomain on the expression and axonal localization of the Robo3 protein. In wild type embryos, Robo3 protein is detectable on axons in the ventral nerve cord, and restricted to longitudinal axons in the lateral two-thirds of the neuropile (the intermediate and lateral zones), i.e. Robo3-expressing axons are excluded from the medial zone. Like Robo1 and Robo2, Robo3 protein is excluded from commissural (midline-crossing) axon segments and very little if any Robo3 protein is detectable on commissures. HA-tagged full-length Robo3 protein expressed from the modified robo3 robo3 allele fully reproduces this expression pattern. In embryos homozygous for the robo3δC allele, Robo3δC protein is not restricted to the intermediazte and lateral zones, and is instead detectable at uniform levels across the entire width of the longitudinal connectives. In addition, Robo3δC protein is not excluded from commissures, and instead is detectable on midline-crossing and non-crossing axons alike in robo3δC embryos. These differences in protein expression appear to be due at least in part to misguidance of Robo3-expressing axons, as embryos that are heterozygous for the robo3δC allele (robo3δC /+) exhibit proper exclusion of Robo3δC protein from the medial zone and much lower levels of Robo3δC protein on commissures (though still slightly elevated compared to wild type and robo3 robo3 embryos). The presence of Robo3δC protein on axons in the medial zone is interpreted as intermediate or lateral Robo3-positive axons inappropriately entering the medial zone, rather than inappropriate expression of Robo3δC protein on normally Robo3-negative medial axons. Consistent with this interpretation, below is shown that FasII-positive intermediate longitudinal axons are also misguided into the medial zone in robo3δC embryos. A similar expansion of Robo3-positive axons into the medial zone has recently been reported in embryos expressing a version of Robo3 from which the Slit-binding Ig1 domain is deleted (robo3δIg1 ) (Agcaoili, 2024).
Next, the axon guidance activity of Robo3δC was examined by using anti-FasII antibody to label distinct longitudinal axon tracts in the medial, intermediate, and lateral zones of the ventral nerve cord. In wild type embryos or embryos homozygous for the robo3 allele, distinct FasII-positive axon pathways form in each of the three zones. In robo3 In addition to longitudinal pathway defects, some FasII-positive medial axons ectopically cross the midline in robo3δC embryos, a phenotype that is not observed in robo31 mutants. It is speculated that this may be caused by a dominant-negative-like effect of Robo3δC expression on Robo1 and/or Robo2, both of which are required for midline repulsion and both of which are expected to be co-expressed with Robo3 in some neurons. A similar effect can be induced through pan-neuronal misexpression of Robo1δC or Robo2δC. Ectopic midline crossing of some Robo3δC-expressing neurons would also account for the presence of Robo3δC protein on commissural axon segments (Agcaoili, 2024).
These results demonstrate that Robo3's ability to guide intermediate longitudinal axons is reduced, but not eliminated, by the loss of its cytoplasmic domain. Thus, any downstream signaling events that are mediated directly by the Robo3 cytodomain are not absolutely required for its role in longitudinal axon guidance. The exact mechanism by which Robo3 guides longitudinal axons is not yet known, or what other factor(s) may be involved. It is possible, for example, that Robo3 could function in a heteromeric signaling complex with one or more co-receptors, which could compensate for the loss of Robo3 cytodomain-dependent signaling. Our results therefore do not necessarily rule out the possibility that a Robo3-dependent signaling pathway is necessary for guidance of intermediate longitudinal axons, but instead only show that the Robo3 cytodomain itself is not strictly required.The results further indicate that some sequences within the Robo3 cytodomain do contribute to its longitudinal axon guidance role, since the robo3δC allele displays reduced function. One possibility is that the reduced activity of robo3δC is due to loss of the evolutionarily conserved CC0 and CC1 sequence motifs. The CC0 and CC1 sequences are also conserved in Drosophila Robo1 and Robo2, which can both substitute for Robo3 to promote intermediate pathway formation. Further studies, for example smaller deletions targeting subregions within the cytodomain or engineered point mutations targeting hypothesized phosphorylation sites, will be necessary to precisely define the functional contributions of specific cytodomain sequences in Robo3 (Agcaoili, 2024).
Mutations in ten-eleven translocation (TET) proteins are associated with human neurodevelopmental disorders. This study found a function of Tet in regulating Drosophila early brain development. The Tet DNA-binding domain TetAXXC is required for axon guidance in the mushroom body (MB). Glutamine synthetase 2 (Gs2), a key enzyme in glutamatergic signaling, is significantly down-regulated in the TetAXXC brains. Loss of Gs2 recapitulates the TetAXXC phenotype. Surprisingly, Tet and Gs2 act in the insulin-producing cells (IPCs) to control MB axon guidance, and overexpression of Gs2 in IPCs rescues the defects of TetAXXC. Feeding TetAXXC with metabotropic glutamate receptor antagonist MPEP rescues the phenotype while glutamate enhances it. Mutants in Tet and Drosophila Fmr1, the homolog of human FMR1, have similar defects, and overexpression of Gs2 in IPCs also rescues the Fmr1 phenotype. This study provides the first evidence that Tet controls the guidance of developing brain axons by modulating glutamatergic signaling (Tran, 2024).
TET is essential for neurodevelopment, but how it functions in the developing brain is not well understood. For instance, TET3 is upregulated and required for axon regeneration in adult dorsal root ganglion (DRG) neurons after injury, and Tet is involved in neuronal functions including commissure axon patterning and glial homeostasis in Drosophila. However, the requirement of Tet and its DNA-binding domain in regulating axon guidance in the developing brain has not been studied. The current results establish that Tet regulates the guidance of β axons in the MB, a prominent structure in the brain that controls different neurological functions, and this regulation is dependent on its DNA-binding domain (Tran, 2024).
The results also show that the requirement for Tet function in the MB β axon guidance is limited to the first 48 h of pupal brain development, at a time when β axons grow to form the mature MB β lobes. When Tet levels are reduced β axons are not restricted to one hemisphere, rather they grow across the brain midline. Careful inspection of the Tet RNAi axons that cross the midline showed that these axons formed synaptic boutons in the "wrong" brain hemisphere suggesting that the circuits of the hemispheres are misconnected. Together these observations suggest that treatment for neurodevelopmental disorders that are due to axon guidance problems and the ensuing misconnection of synapses needs to be applied at early stages of nervous system development (Tran, 2024).
Transcriptomic studies demonstrated that Glutamine synthetase 2 (Gs2) and GABA transporter (Gat) are generally regulated by Tet in the brain, but this study concentrated on their requirement in the MB. Gs2 is a key enzyme in regulating glutamate levels in glutamatergic signaling and Gat is the neurotransporter that controls GABA levels in the GABAergic signaling suggesting that Tet is required in both pathways. However, knockdown of Gat did not result in an β lobe phenotype suggesting that Gat is not required for normal axon guidance. In contrast, inducing Gs2 RNAi by the 201Y-GAL4 driver resulted in axon defects suggesting that MB axon guidance is regulated by glutamatergic signaling via Gs2. In addition, treating TetAXXC mutants with MPEP, a metabotropic glutamate receptor (mGluR) antagonist, can partially rescue the axon guidance defects while feeding mutants glutamate enhances the TetAXXC phenotype. These data demonstrate a new function of Tet in regulating glutamatergic signaling in the brain and support the notion that the pathway is mis-regulated in Tet mutant brains. Glutamatergic signaling is a major excitatory signal in the human brain and the pathway is also known to act in neural developmental events such as proliferation, differentiation, migration, and synaptogenesis. Interestingly, elevated levels of glutamatergic signaling have also been proposed as the underlying mechanism of the neurodevelopmental disorder in FXS.50 Based on the conserved functional domains of Tet in humans and flies, the mis-regulation of the glutamatergic signaling observed in Drosophila brain during early development may also occur in developing human brains harboring Tet mutations that lead to neurodevelopmental disorders (Tran, 2024).
Previousl work has mapped Tet-DNA -binding sites in the genome by ChIP-seq and mapped Tet-mediated 5hmrC RNA modification throughout the transcriptome by hMeRIP-seq. There is no Tet binding detected by ChIP-seq on the gene body or promoter region of Gs2 and Gat. In addition, the Gs2 and Gat mRNAs do not carry the Tet-dependent 5hmrC modification. These observations suggest that Tet regulates Gs2 and Gat indirectly. This study investigated if robo2/slit, which were identified as RNA modification targets of Tet, could regulate MB axon guidance. However, no significant effects were observed on MB axon guidance upon robo2 knockdown in the MB. Therefore, it is proposed that one or several Tet target genes are responsible for controlling the transcription or RNA levels of Gs2 and Gat (Tran, 2024).
Alternatively, Tet may have a novel, so far unknown function. Since Gs2 and Gat are also down-regulated in the Fmr13 mutant, Tet and Fmr1 may function together to control the mRNA levels of these two and potentially additional genes. Fmr1 was previously found in the regulatory protein complex Not1/CCR4 which is known to control all steps of gene expression from transcription in the nucleus to mRNA degradation in the cytoplasm depending on the composition of proteins that interact with the complex. In this case the DNA-binding domain of Tet may be responsible for protein-protein interaction(s) necessary to form the complex. Previous work has identified Fmr1 in a complex with the zinc finger protein RP-8 (Zfrp8), which functions in the assembly of mRNA ribonucleoprotein (mRNP) complexes. Interestingly, by co-immunoprecipitation this study found Zfrp8 and Tet in the same complex. Thus, Tet and Fmr1 may both coordinate with the Not1/CCR4 or other mRNP complexes to regulate the expression levels of Gs2 and Gat. Indeed, understanding these complexes and their functions in controlling mRNA levels is of great importance, especially since RNA processing is not well understood (Tran, 2024).
IPCs in Drosophila are a group of neurons located at the dorsal midline of the central brain that produce and secrete insulin-like peptides into the hemolymph to regulate growth and metabolism. Previous studies have shown that IPCs can also regulate neuronal functions. For instance, overexpressing Fmr1 in IPCs could rescue the circadian defects observed in Fmr1 mutants. But the involvement of IPCs in regulating MB axon guidance has not previously been shown. In RNA-seq experiments, Gs2 is the most significantly reduced transcript in the Tet>AXXC mutant brain. Gs2 is expressed in the IPCs but not in MB neurons. Tet is also expressed in IPCs and in agreement with this expression the axon midline-crossing phenotype is detected when either Tet or Gs2 in IPCs specifically knocked down. In addition, the axon midline-crossing phenotype was rescued when expressing Gs2 in the IPCs of Tet and Fmr1 mutants. Hence, the results point to a previously unknown function of IPCs in regulating MB axon guidance via glutamatergic signaling in the developing brain (Tran, 2024).
In addition to IPCs, Gs2 is also expressed in glial cells where it is responsible for converting glutamate to glutamine to regulate the levels of the neurotransmitter glutamate in the brain. Thus, the reduction of Gs2 levels in TetAXXC mutants may lead to the accumulation of glutamate and a deficiency of glutamine in the IPCs. However, the mechanism by which Tet and Gs2 regulate axon guidance via IPCs needs to be further elucidated. One possible mechanism is that in Tet mutant brains Gs2 is reduced in the IPCs leading to the accumulation of glutamate in the IPCs that is then secreted at the brain midline. The elevated levels of glutamate at the midline could cause MB β axons to cross over the midline. In support of this possible mechanism, glutamate has been reported to reduce the responsiveness of axonal growth cones to repellent cues such as Slit-2 and Sema-3A/C23 and also to enhance the growth rate and branching of dopaminergic axons. In addition, glutamate can be released by pancreatic cells, which are the insulin-secreting cells in mammals, via excitatory amino acid transporters (EAAT) by uptake reversal. The uptake reversal activity of EAAT has also been reported in astrocytes and it depends on the relative intracellular and extracellular glutamate concentration. Further studies are needed to test the secretion of glutamate by IPCs and to investigate whether the MB β axon crossing phenotype is due to elevated glutamate levels at the midline of TetAXXC mutant brains (Tran, 2024).
Previously, the glutamate receptor antagonist MPEP was successfully used to suppress the axonal defect in Fmr13 mutants while high glutamate concentration can enhance the phenotype. The observation that MPEP and glutamate have similar effects on the TetAXXC phenotype suggests that Tet and Fmr1 have overlapping functions. This conclusion is further strengthened by the finding that Gs2 is down-regulated in the brain of both TetAXXC and Fmr13 mutants and that increasing Gs2 levels by overexpressing it from a transgene reduced the occurrence of the β lobe fusion phenotype in both TetAXXC and Fmr13 mutants. The rescue is partial, suggesting that the expression levels of Gs2 are essential additional targets of Tet and Fmr1 may also contribute to the phenotype. As Tet and Fmr1 can regulate a broad spectrum of genes, it is impressive that overexpression of only one gene, Gs2, encoding a key enzyme in the glutamatergic pathway, can rescue the axonal defect in about half of both Tet and Fmr1 mutants (Tran, 2024).
Dysfunction of glutamatergic signaling is involved in a variety of human diseases including FXS, autism spectrum disorder, schizophrenia, neurodegenerative diseases, and gliomas. The current results suggest that glutamine synthetase is a potential target for early intervention. Recently, an increasing number of studies have shown that Tet mutations are associated with neurodevelopmental disorders in humans. The mechanism by which human TET mutations cause these disorders needs to be elucidated, but the highly conserved functional domains of human and Drosophila Tet suggest that the Tet function in axon guidance that was found in Drosophila may be conserved in humans. Thus, this study provides initial evidence that glutamatergic signaling is altered in the Tet mutant brains and the mutant may serve as a Drosophila model for drug screening and study of neurodevelopmental disorders caused by TET mutations (Tran, 2024).
This study described the new function of Tet in controlling axon guidance in the Drosophila brain by regulating glutamatergic signaling via Gs2. Tet was shown to indirectly regulates Gs2 levels and suggest a possible underlying mechanism. However further studies need to be conducted to confirm how Tet regulates Gs2 transcript levels. This study also demonstrated that Tet and Gs2 are required in the IPCs to control axon guidance. How glutamate is secreted from IPCs and becomes localized to the brain midline and how the outgrowth of the MB axons is misguided by excess glutamate is not known. Identifying a transporter (e.g., EAAT) in the IPCs that is responsible for the secretion of glutamate and a receptor on the MB axons that is responsible for the guidance is necessary to understand the mechanism of how the IPCs regulate MB axons via the neurotransmitter glutamate (Tran, 2024).
A navigating axon faces complex choices when selecting postsynaptic partners in a three-dimensional (3D) space. In this work a principle was discovered that can establish the 3D glomerular map of the fly antennal lobe by reducing the higher dimensionality serially to 1D projections. During development, olfactory receptor neuron (ORN) axons first contact their partner projection neuron dendrites on the spherical surface of the antennal lobe, regardless of whether the adult glomeruli lie near the surface or inside. Along this 2D surface, axons of each ORN type take a specific, arc-shaped trajectory that precisely intersects with their partner dendrites. Altering axon trajectories compromises synaptic partner matching. A 3D search is thus reduced to one dimension, simplifying partner matching (Lyu, 2025).
References Abe, T., Yamazaki, D., Murakami, S., Hiroi, M., Nitta, Y., Maeyama, Y. and Tabata, T. (2014). The NAV2 homolog Sickie regulates F-actin-mediated axonal growth in Drosophila mushroom body neurons via the non-canonical Rac-Cofilin pathway. Development 141: 4716-4728. PubMed ID: 25411210
Agcaoili, J., Evans, T. A. (2024). Drosophila Robo3 guides longitudinal axons partially independently of its cytodomain. microPublication biology, 2024 PubMed ID: 38882930
Apitz, H. and Salecker, I. (2018). Spatio-temporal relays control layer identity of direction-selective neuron subtypes in Drosophila. Nat Commun 9(1): 2295. PubMed ID: 29895891
Avery, M. A., Rooney, T. M., Pandya, J. D., Wishart, T. M., Gillingwater, T. H., Geddes, J. W., Sullivan, P. G. and Freeman, M. R. (2012). WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol 22: 596-600. PubMed ID: 22425157
Banerjee, S. and Riordan, M. (2018). Coordinated regulation of axonal microtubule organization and transport by Drosophila Neurexin and BMP pathway. Sci Rep 8(1): 17337. PubMed ID: 30478335
Banka, S., Bennington, A., ..., Kazanietz, M. G. and Millard, T. H. (2022). Activating RAC1 variants in the switch II region cause a developmental syndrome and alter neuronal morphology. Brain. PubMed ID: 35139179
Barish, S., Nuss, S., Strunilin, I., Bao, S., Mukherjee, S., Jones, C. D. and Volkan, P. C. (2018). Combinations of DIPs and Dprs control organization of olfactory receptor neuron terminals in Drosophila. PLoS Genet 14(8): e1007560. PubMed ID: 30102700
Boyan, G., Williams, L., Ehrhardt, E. (2023). Central projections from Johnston's organ in the locust: Axogenesis and brain neuroarchitecture. Dev Genes Evol, 233(2):147-159 PubMed ID: 37695323
Brierley, D. J., Blanc, E., Reddy, O. V., Vijayraghavan, K. and Williams, D. W. (2009). Dendritic targeting in the leg neuropil of Drosophila: the role of midline signalling molecules in generating a myotopic map. PLoS Biol 7(9): e1000199. PubMed ID: 19771147
Carranza, A., Howard, L. J., Brown, H. E., Ametepe, A. S. and Evans, T. A. (2023). Slit-independent guidance of longitudinal axons by Drosophila Robo3. bioRxiv. PubMed ID: 37214810
Carrillo, R. A., Ozkan, E., Menon, K. P., Nagarkar-Jaiswal, S., Lee, P. T., Jeon, M., Birnbaum, M. E., Bellen, H. J., Garcia, K. C. and Zinn, K. (2015). Control of Synaptic Connectivity by a Network of Drosophila IgSF Cell Surface Proteins. Cell 163(7): 1770-1782. PubMed ID: 26687361
Catela, C., Shin, M. M. and Dasen, J. S. (2015). Assembly and function of spinal circuits for motor control. Annu Rev Cell Dev Biol 31: 669-698. PubMed ID: 26393773
Contreras, E. G., Palominos, T., Glavic, A., Brand, A. H., Sierralta, J. and Oliva, C. (2018). The transcription factor SoxD controls neuronal guidance in the Drosophila visual system. Sci Rep 8(1): 13332. PubMed ID: 30190506
Corty, M. M., Hulegaard, A. L., Hill, J. Q., Sheehan, A. E., Aicher, S. A. and Freeman, M. R. (2022). Discoidin domain receptor regulates ensheathment, survival, and caliber of peripheral axons. Development. PubMed ID: 36355066
Del Castillo, U., Lu, W., Winding, M., Lakonishok, M. and Gelfand, V. I. (2014). Pavarotti/MKLP1 regulates microtubule sliding and neurite outgrowth in Drosophila neurons. Curr Biol 25(2):200-5. PubMed ID: 25557664
Del Castillo, U., Muller, H. J. and Gelfand, V. I. (2020). Kinetochore protein Spindly controls microtubule polarity in Drosophila axons. Proc Natl Acad Sci U S A 117(22): 12155-12163. PubMed ID: 32430325
Desai, K. K., Cheng, C. L., Bingman, C. A., Phillips, G. N., Jr. and Raines, R. T. (2014). A tRNA splicing operon: Archease endows RtcB with dual GTP/ATP cofactor specificity and accelerates RNA ligation. Nucleic Acids Res 42: 3931-3942. PubMed ID: 24435797
Enneking, E. M., Kudumala, S. R., Moreno, E., Stephan, R., Boerner, J., Godenschwege, T. A. and Pielage, J. (2013). Transsynaptic coordination of synaptic growth, function, and stability by the L1-type CAM Neuroglian. PLoS Biol 11(4): e1001537. PubMed ID: 23610557
Evans, T. A., Bashaw, G. J. (2010) Functional diversity of Robo receptor immunoglobulin domains promotes distinct axon guidance decisions. Curr Biol 20: 567-572. PubMed ID: 20206526
Fang, H. Y., Forghani, R., Clarke, A., McQueen, P. G., Chandrasekaran, A., O'Neill, K. M., Losert, W., Papoian, G. A. and Giniger, E. (2023). Enabled primarily controls filopodial morphology, not actin organization, in the TSM1 growth cone in Drosophila. Mol Biol Cell 34(8): ar83. PubMed ID: 37223966
Fang, Y., Soares, L., Teng, X., Geary, M. and Bonini, N. M. (2012). A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr Biol 22: 590-595. PubMed ID: 22425156
Foldi, I., Toth, K., Gombos, R., Gaszler, P., Gorog, P., Zygouras, I., Bugyi, B. and Mihaly, J. (2022). Molecular Dissection of DAAM Function during Axon Growth in Drosophila Embryonic Neurons. Cells 11(9). PubMed ID: 35563792
Foley, E. and O'Farrell, P. H. (2004). Functional dissection of an innate immune response by a genome-wide RNAi screen. PLoS Biol 2: E203. PubMed ID: 15221030
Genschik, P., Billy, E., Swianiewicz, M. and Filipowicz, W. (1997). The human RNA 3'-terminal phosphate cyclase is a member of a new family of proteins conserved in Eucarya, Bacteria and Archaea. EMBO J 16: 2955-2967. PubMed ID: 9184239
Gu, T., Zhao, T., Kohli, U. and Hewes, R. S. (2017). The large and small SPEN family proteins stimulate axon outgrowth during neurosecretory cell remodeling in Drosophila. Dev Biol 431(2):226-238. PubMed ID: 28916169
Hahn, I., Voelzmann, A., Parkin, J., Fulle, J. B., Slater, P. G., Lowery, L. A., Sanchez-Soriano, N. and Prokop, A. (2021). Tau, XMAP215/Msps and Eb1 co-operate interdependently to regulate microtubule polymerisation and bundle formation in axons. PLoS Genet 17(7): e1009647. PubMed ID: 34228717
Hsu, J. M., Kang, Y., Corty, M. M., Mathieson, D., Peters, O. M. and Freeman, M. R. (2020). Injury-induced inhibition of bystander neurons requires dSarm and signaling from glia. Neuron. PubMed ID: 33296670
Hu, Y., Park, K. K., Yang, L., Wei, X., Yang, Q., Cho, K. S., Thielen, P., Lee, A. H., Cartoni, R., Glimcher, L. H., Chen, D. F. and He, Z. (2012). Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron 73: 445-452. PubMed ID: 22325198
Ichinose, S., Ogawa, T., Jiang, X. and Hirokawa, N. (2019). The spatiotemporal construction of the axon initial segment via KIF3/KAP3/TRIM46 transport under MARK2 signaling. Cell Rep 28(9): 2413-2426. PubMed ID: 31461655
Issman-Zecharya, N. and Schuldiner, O. (2014). The PI3K class III
complex promotes axon pruning by downregulating a Ptc-derived signal via
endosome-lysosomal degradation. Dev Cell 31: 461-473. PubMed ID: 25458013
Izadifar, A., Courchet, J., Virga, D. M., Verreet, T., Hamilton, S., Ayaz, D., Misbaer, A., Vandenbogaerde, S., Monteiro, L., Petrovic, M., Sachse, S., Yan, B., Erfurth, M. L., Dascenco, D., Kise, Y., Yan, J., Edwards-Faret, G., Lewis, T., Polleux, F. and Schmucker, D. (2021). Axon morphogenesis and maintenance require an evolutionary conserved safeguard function of Wnk kinases antagonizing Sarm and Axed. Neuron. PubMed ID: 34384519
Jakobs, M. A. H., Zemel, A. and Franze, K. (2022). Unrestrained growth of correctly oriented microtubules instructs axonal microtubule orientation. Elife 11. PubMed ID: 36214669
Jegla, T., Nguyen, M. M., Feng, C., Goetschius, D. J., Luna, E., van Rossum, D. B., Kamel, B., Pisupati, A., Milner, E. S. and Rolls, M. M. (2016). Bilaterian giant Ankyrins have a common evolutionary origin and play a conserved role in patterning the axon initial segment. PLoS Genet 12(12): e1006457. PubMed ID: 27911898
Ji, W., Wu, L. F. and Altschuler, S. J. (2021). Analysis of growth cone extension in standardized coordinates highlights self-organization rules during wiring of the Drosophila visual system. PLoS Genet 17(11): e1009857. PubMed ID: 34731164
Jurkin, J., Henkel, T., Nielsen, A. F., Minnich, M., Popow, J., Kaufmann, T., Heindl, K., Hoffmann, T., Busslinger, M. and Martinez, J. (2014). The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J 33: 2922-2936. PubMed ID: 25378478
Kelly, S. M., Bienkowski, R., Banerjee, A., Melicharek, D. J., Brewer, Z. A., Marenda, D. R., Corbett, A. H. and Moberg, K. H. (2016). The Drosophila ortholog of the Zc3h14 RNA binding protein acts within neurons to pattern axon projection in the developing brain. Dev Neurobiol 76(1): 93-106. PubMed ID: 25980665
Kinold, J. C., Pfarr, C. and Aberle, H. (2018). Sidestep-induced neuromuscular miswiring causes severe locomotion defects in Drosophila larvae. Development 145(17). PubMed ID: 30166331
Kurmangaliyev, Y. Z., Yoo, J., LoCascio, S. A. and Zipursky, S. L. (2019). Modular transcriptional programs separately define axon and dendrite connectivity. Elife 8. PubMed ID: 31687928
Lacoste, J., Soula, H., Burg, A., Audibert, A., Darnat, P., Gho, M. and Louvet-Vallee, S. (2022). A neural progenitor mitotic wave is required for asynchronous axon outgrowth and morphology. Elife 11. PubMed ID: 35254258
Landgraf, M., et al. (2003). Embryonic origins of a motor system: Motor dendrites form a myotopic map in Drosophila.
PLoS Biol. 1(2):E41. PubMed ID: 14624243
Lavergne, G., Zmojdzian, M., Da Ponte, J. P., Junion, G. and Jagla, K. (2020). Drosophila adult muscle precursor cells contribute to motor axon pathfinding and proper innervation of embryonic muscles. Development 147(4). PubMed ID: 32001438
Li, H., Watson, A., Olechwier, A., Anaya, M., Sorooshyari, S. K., Harnett, D. P., Lee, H. P., Vielmetter, J., Fares, M. A., Garcia, K. C., Ozkan, E., Labrador, J. P. and Zinn, K. (2017). Deconstruction of the beaten Path-Sidestep interaction network provides insights into neuromuscular system development. Elife 6. PubMed ID: 28829740
Lobb-Rabe, M., DeLong, K., Salazar, R. J., Zhang, R., Wang, Y. and Carrillo, R. A. (2022). Dpr10 and Nocte are required for Drosophila motor axon pathfinding. Neural Dev 17(1): 10. PubMed ID: 36271407
Long, D. R., Kinser, A., Olalde-Welling, A., Brewer, L., Lim, J., Matheny, D., Long, B., Roossien, D. H. (2023). 5-HT1A regulates axon outgrowth in a subpopulation of Drosophila serotonergic neurons. Developmental neurobiology. 83(7-8):268-281 PubMed ID: 37714743
Lu, Y., Belin, S. and He, Z. (2014). Signaling regulations of neuronal regenerative ability. Curr Opin Neurobiol 27: 135-142. PubMed ID: 24727245
Maiya, R., Dey, S., Ray, K. and Menon, G. I. (2023). The interplay of active and passive mechanisms in slow axonal transport. Biophys J 122(2): 333-345. PubMed ID: 36502274
Malartre, M., Ayaz, D., Amador, F. F. and Martin-Bermudo, M. D. (2010). The guanine exchange factor Vav controls axon growth and guidance during Drosophila development. J Neurosci 30: 2257-2267. PubMed ID: 20147552
Martin-Pena, A. and Ferrus, A. (2019). CCB is involved in actin-based axonal transport of selected synaptic proteins. J Neurosci. PubMed ID: 31754011
Mauss, A., Tripodi, M., Evers, J. F. and Landgraf, M. (2009). Midline signalling systems direct the formation of a neural map by dendritic targeting in the Drosophila motor system. PLoS Biol 7(9): e1000200. PubMed ID: 19771146
Mino, R. E., Rogers, S. L., Risinger, A. L., Rohena, C., Banerjee, S. and Bhat, M. A. (2016). Drosophila Ringmaker regulates microtubule stabilization and axonal extension during embryonic development. J Cell Sci [Epub ahead of print]. PubMed ID: 27422099
Monahan Vargas, E. J., Matamoros, A. J., Qiu, J., Jan, C. H., Wang, Q., Gorczyca, D., Han, T. W., Weissman, J. S., Jan, Y. N., Banerjee, S. and Song, Y. (2020). The microtubule regulator ringer functions downstream from the RNA repair/splicing pathway to promote axon regeneration. Genes Dev 34(3-4): 194-208. PubMed ID: 31919191
Neukomm, L. J., Burdett, T. C., Seeds, A. M., Hampel, S., Coutinho-Budd, J. C., Farley, J. E., Wong, J., Karadeniz, Y. B., Osterloh, J. M., Sheehan, A. E. and Freeman, M. R. (2017). Axon death pathways converge on Axundead to promote functional and structural axon disassembly. Neuron 95(1): 78-91.e75. PubMed ID: 28683272
Neukomm, L. J., Burdett, T. C., Seeds, A. M., Hampel, S., Coutinho-Budd, J. C., Farley, J. E., Wong, J., Karadeniz, Y. B., Osterloh, J. M., Sheehan, A. E. and Freeman, M. R. (2017). Axon death pathways converge on Axundead to promote functional and structural axon disassembly. Neuron 95(1): 78-91.e75. PubMed ID: 28683272
Nguyen, C. T., Nguyen, V. M. and Jeong, S. (2022). Regulation of Off-track bidirectional signaling by Semaphorin-1a and Wnt signaling in the Drosophila motor axon guidance. Insect Biochem Mol Biol 150: 103857. PubMed ID: 36244650
Nitta, Y., Yamazaki, D., Sugie, A., Hiroi, M. and Tabata, T. (2016). DISCO Interacting Protein 2 regulates axonal bifurcation and guidance of Drosophila mushroom body neurons. Dev Biol [Epub ahead of print]. PubMed ID: 27908785
Nix, P., Hammarlund, M., Hauth, L., Lachnit, M., Jorgensen, E. M. and Bastiani, M. (2014). Axon regeneration genes identified by RNAi screening in C. elegans. J Neurosci 34: 629-645. PubMed ID: 24403161
Ozkan, A. and Berberoglu, H. (2013). Cell to substratum and cell to cell interactions of microalgae. Colloids Surf B Biointerfaces 112: 302-309. PubMed ID: 24004676
Pappu, K. S., Morey, M., Nern, A., Spitzweck, B., Dickson, B. J., Zipursky, S. L. (2011). Robo-3-mediated repulsive interactions guide R8 axons during Drosophila visual system development. Proc Natl Acad Sci U S A 108: 7571-7576. PubMed ID: 21490297
Pielage, J., Cheng, L., Fetter, R. D., Carlton, P. M., Sedat, J. W. and Davis, G. W. (2008). A presynaptic giant ankyrin stabilizes the NMJ through regulation of presynaptic microtubules and transsynaptic cell adhesion. Neuron 58(2): 195-209. PubMed ID: 18439405
Pinto-Teixeira, F., Koo, C., Rossi, A. M., Neriec, N., Bertet, C., Li, X., Del-Valle-Rodriguez, A. and Desplan, C. (2018). Development of concurrent retinotopic maps in the fly motion detection circuit. Cell 173(2): 485-498 e411. PubMed ID: 29576455
Pollitt, S. L., Myers, K. R., Yoo, J. and Zheng, J. Q. (2020). LIM and SH3 Protein 1 Localizes to the Leading Edge of Protruding Lamellipodia and Regulates Axon Development. Mol Biol Cell: mbcE20060366. PubMed ID: 32997597
Popow, J., Englert, M., Weitzer, S., Schleiffer, A., Mierzwa, B., Mechtler, K., Trowitzsch, S., Will, C. L., Luhrmann, R., Soll, D. and Martinez, J. (2011). HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 331: 760-764. PubMed ID: 21311021
Popow, J., Jurkin, J., Schleiffer, A. and Martinez, J. (2014). Analysis of orthologous groups reveals archease and DDX1 as tRNA splicing factors. Nature 511: 104-107. PubMed ID: 24870230
Qu, Y., Hahn, I., Lees, M., Parkin, J., Voelzmann, A., Dorey, K., Rathbone, A., Friel, C. T., Allan, V. J., Okenve-Ramos, P., Sanchez-Soriano, N. and Prokop, A. (2019). Efa6 protects axons and regulates their growth and branching by inhibiting microtubule polymerisation at the cortex. Elife 8. PubMed ID: 31718774
Razetti, A., Medioni, C., Malandain, G., Besse, F. and Descombes, X. (2018). A stochastic framework to model axon interactions within growing neuronal populations. PLoS Comput Biol 14(12): e1006627. PubMed ID: 30507939
Remus, B. S. and Shuman, S. (2013). A kinetic framework for tRNA ligase and enforcement of a 2'-phosphate requirement for ligation highlights the design logic of an RNA repair machine. RNA 19: 659-669. PubMed ID: 23515942
Roblodowski, C. and He, Q. (2017). Drosophila Dunc-115 mediates axon projection through actin binding. Invert Neurosci 17(1): 2. PubMed ID: 28124181
Ron, D. and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519-529. PubMed ID: 17565364
Roossien, D. H., Lamoureux, P., Van Vactor, D. and Miller, K. E. (2013). Drosophila growth cones advance by forward translocation of the neuronal cytoskeletal meshwork in vivo. PLoS One 8: e80136. PubMed ID: 24244629
Ryoo, H. D., Domingos, P. M., Kang, M. J. and Steller, H. (2007). Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J 26: 242-252. PubMed ID: 17170705
Sakuma, C., Kawauchi, T., Haraguchi, S., Shikanai, M., Yamaguchi, Y., Gelfand, V. I., Luo, L., Miura, M. and Chihara, T. (2014). Drosophila Strip serves as a platform for early endosome organization during axon elongation. Nat Commun 5: 5180. PubMed ID: 25312435
Santiago, C., Labrador, J. P., Bashaw, G. J. (2014) The Homeodomain Transcription Factor Hb9 Controls Axon Guidance in Drosophila through the Regulation of Robo Receptors. Cell Rep 7: 153-165. PubMed ID: 24685136
Santiago, C. and Bashaw, G.J. (2017). Islet coordinately regulates motor axon guidance and dendrite
targeting through the Frazzled/DCC receptor. Cell Rep 18: 1646-1659. PubMed ID: 28199838
Song, Y., Ori-McKenney, K. M., Zheng, Y., Han, C., Jan, L. Y. and Jan, Y. N. (2012). Regeneration of Drosophila sensory neuron axons and dendrites is regulated by the Akt pathway involving Pten and microRNA bantam. Genes Dev 26: 1612-1625. PubMed ID: 22759636
Song, Y., Sretavan, D., Salegio, E. A., Berg, J., Huang, X., Cheng, T., Xiong, X., Meltzer, S., Han, C., Nguyen, T. T., Bresnahan, J. C., Beattie, M. S., Jan, L. Y. and Jan, Y. N. (2015). Regulation of axon regeneration by the RNA repair and splicing pathway. Nat Neurosci 18: 817-825. PubMed ID: 25961792
Spindler, S. R., Ortiz, I., Fung, S., Takashima, S. and Hartenstein, V. (2009). Drosophila cortex and neuropile glia influence secondary axon tract growth, pathfinding, and fasciculation in the developing larval brain.
Dev. Biol. 334(2): 355-68. PubMed ID: 19646433
Spitzweck, B., Brankatschk, M., Dickson, B. J. (2010) Distinct protein domains and expression patterns confer divergent axon guidance functions for Drosophila Robo receptors. Cell 140: 409-420. PubMed ID: 20144763
Spurrier, J., Shukla, A. K., Buckley, T., Smith-Trunova, S., Kuzina, I., Gu, Q. and Giniger, E. (2019). Expression of a fragment of Ankyrin 2 disrupts the structure of the axon initial segment and causes axonal degeneration in Drosophila. Mol Neurobiol. PubMed ID: 30666562
Stephan, R., Goellner, B., Moreno, E., Frank, C. A., Hugenschmidt, T., Genoud, C., Aberle, H. and Pielage, J. (2015). Hierarchical microtubule organization controls axon caliber and transport and determines synaptic structure and stability. Dev Cell 33(1): 5-21. PubMed ID: 25800091
Stone, M. C., Mauger, A. S. and Rolls, M. M. (2023). Ciliated sensory neurons can regenerate axons after complete axon removal. J Exp Biol 226(12). PubMed ID: 37212026
Sung, H. and Lloyd, T. E. (2022). Defective axonal transport of endo-lysosomes and dense core vesicles in a Drosophila model of C9-ALS/FTD. Traffic 23(9): 430-441. PubMed ID: 35908282
Swope, R. D., Hertzler, J. I., Stone, M. C., Kothe, G. O. and Rolls, M. M. (2022). The exocyst complex is required for developmental and regenerative neurite growth in vivo. Dev Biol 492: 1-13. PubMed ID: 36162553
Syed, D. S., Gowda, S. B., Reddy, O. V., Reichert, H. and VijayRaghavan, K. (2016). Glial and neuronal Semaphorin signaling instruct the development of a functional myotopic map for Drosophila walking. Elife 5: e11572. PubMed ID: 26926907
Tan, L., Zhang, K. X., Pecot, M. Y., Nagarkar-Jaiswal, S., Lee, P. T., Takemura, S. Y., McEwen, J. M., Nern, A., Xu, S., Tadros, W., Chen, Z., Zinn, K., Bellen, H. J., Morey, M. and Zipursky, S. L. (2015). Ig Superfamily Ligand and Receptor Pairs Expressed in Synaptic Partners in Drosophila. Cell 163(7): 1756-1769. PubMed ID: 26687360
Tanaka, N. and Shuman, S. (2011). RtcB is the RNA ligase component of an Escherichia coli RNA repair operon. J Biol Chem 286: 7727-7731. PubMed ID: 21224389
Terzi, A., Roeder, H., Weaver, C. J. and Suter, D. M. (2020). Neuronal NADPH oxidase 2 regulates growth cone guidance downstream of slit2/robo2. Dev Neurobiol. PubMed ID: 33191581
Tran, H., Le, L., Singh, B. N., Kramer, J., Steward, R. (2024). Tet controls axon guidance in early brain development through glutamatergic signaling. iScience, 27(5):109634 PubMed ID: 38655199
Trunova, S., Baek, B. and Giniger, E. (2011). Cdk5 regulates the size of an axon initial segment-like compartment in mushroom body neurons of the Drosophila central brain. J Neurosci 31(29): 10451-10462. PubMed ID: 21775591
Vaikakkara Chithran, A., Allan, D. W. and O'Connor, T. P. (2023). Adult expression of Semaphorins and Plexins is essential for motor neuron survival. Sci Rep 13(1): 5894. PubMed ID: 37041188
Wang, F., Ruppell, K. T., Zhou, S., Qu, Y., Gong, J., Shang, Y., Wu, J., Liu, X., Diao, W., Li, Y. and Xiang, Y. (2023). Gliotransmission and adenosine signaling promote axon regeneration. Dev Cell 58(8): 660-676. PubMed ID: 37028426
Walker, L. J., Summers, D. W., Sasaki, Y., Brace, E. J., Milbrandt, J. and DiAntonio, A. (2017). MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. Elife 6. PubMed ID: 28095293
Weber, T., Stephan, R., Moreno, E. and Pielage, J. (2019). The ankyrin repeat domain controls presynaptic localization of Drosophila Ankyrin2 and is essential for synaptic stability. Front Cell Dev Biol 7: 148. PubMed ID: 31475145
Wolfram, V., Southall, T. D., Gunay, C., Prinz, A. A., Brand, A. H. and Baines, R. A. (2014). The transcription factors islet and Lim3 combinatorially regulate ion channel gene expression. J Neurosci 34(7): 2538-2543. PubMed ID: 24523544
Worpenberg, L., Paolantoni, C., Longhi, S., Mulorz, M. M., Lence, T., Wessels, H. H., Dassi, E., Aiello, G., Sutandy, F. X. R., Scheibe, M., Edupuganti, R. R., Busch, A., Mockel, M. M., Vermeulen, M., Butter, F., Konig, J., Notarangelo, M., Ohler, U., Dieterich, C., Quattrone, A., Soldano, A. and Roignant, J. Y. (2021). Ythdf is a N6-methyladenosine reader that modulates Fmr1 target mRNA selection and restricts axonal growth in Drosophila. Embo J: e104975. PubMed ID: 33428246
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T. and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881-891. PubMed ID: 11779464
Zang, Y., Chaudhari, K. and Bashaw, G. J. (2022). Tace/ADAM17 is a bi-directional regulator of axon guidance that coordinates distinct Frazzled and Dcc receptor signaling outputs. Cell Rep 41(10): 111785. PubMed ID: 36476876
Zhang, Y., Lowe, S., Ding, A. Z. and Li, X. (2023). Notch-dependent binary fate choice regulates the Netrin pathway to control axon guidance of Drosophila visual projection neurons. Cell Rep 42(3): 112143. PubMed ID: 36821442
The Interactive Fly resides on the
Society for Developmental Biology's Web server.
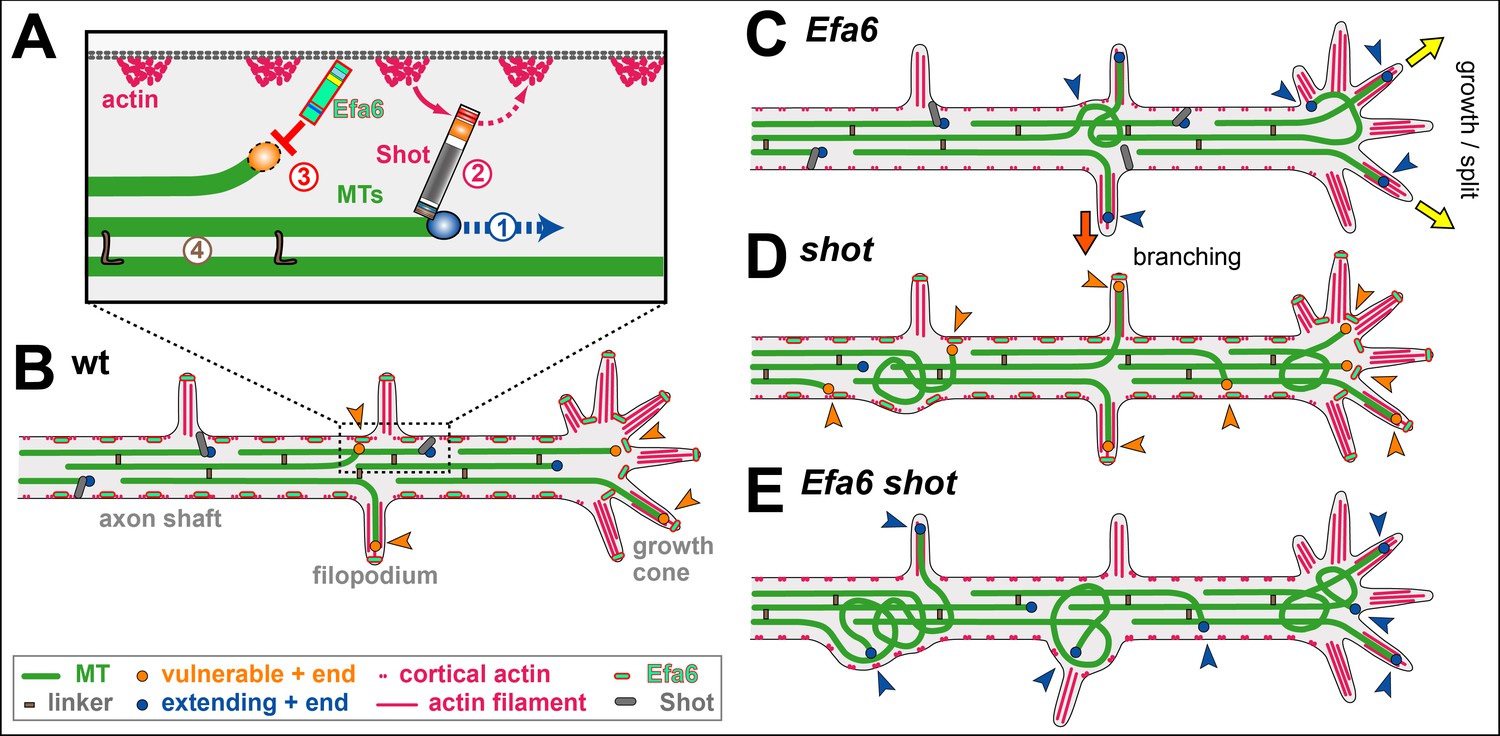{kind=link}
{kind=link}