|
Gene name - Sex combs reduced Synonyms - Cytological map position - 84B1-2 Function - transcription factor Keywords - homeotic - Antennapedia complex, salivary glands |
Symbol - Scr FlyBase ID:FBgn0003339 Genetic map position - 3-47.5 Classification - homeodomain - Antp class (Hoxa5 ortholog) Cellular location - nuclear |
| Recent literature | Li, M., Ma, Z., Liu, J. K., Roy, S., Patel, S. K., Lane, D. C. and Cai, H. N. (2015). An organizational hub of developmentally regulated chromatin loops in the Drosophila Antennapedia complex. Mol Cell Biol 35(23):4018-29. PubMed ID: 26391952
Summary: Sex combs reduced (Scr) is directed by an unusually long regulatory sequence harboring diverse cis elements and an intervening neighbor gene fushi tarazu (ftz). This study reports the presence of a multitude of Chromatin boundary elements (CBEs) in the Scr regulatory region. Selective and dynamic pairing among these CBEs mediates developmentally regulated chromatin loops. In particular, the SF1 boundary plays a central role in organizing two subsets of chromatin loops: one subset encloses ftz, limiting its access by the surrounding Scr enhancers and compartmentalizing distinct histone modifications; and the other subset subdivides the Scr regulatory sequences into independent enhancer access domains. Tandem pairing of SF1 and SF2, two strong CBEs that flank the ftz domain, providing a mechanism for the endogenous Scr enhancer to circumvent the ftz domain. This study demonstrates how an endogenous CBE network, centrally orchestrated by SF1, could remodel the genomic environment to facilitate gene regulation during development. |
Sadasivam, D.A. and Huang, D.H. (2016). Maintenance of tissue pluripotency by epigenetic factors acting at multiple levels. PLoS Genet 12: e1005897. PubMed ID: 26926299 Summary: Pluripotent stem cells often adopt a unique developmental program while retaining certain flexibility. The molecular basis of such properties remains unclear. Using differentiation of pluripotent Drosophila imaginal tissues as assays, this study examined the contribution of epigenetic factors in ectopic activation of Hox genes. It was found that over-expression of Trithorax H3K4 methyltransferase can induce ectopic adult appendages by selectively activating the Hox genes Ultrabithorax and Sex comb reduced in wing and leg discs, respectively. This tissue-specific inducibility correlates with the presence of paused RNA polymerase II in the promoter-proximal region of these genes. Although the Antennapedia promoter is paused in eye-antenna discs, it cannot be induced by Trx without a reduction in histone variants or their chaperones, suggesting additional control by the nucleosomal architecture. Lineage tracing and pulse-chase experiments revealed that the active state of Hox genes is maintained substantially longer in mutants deficient for HIRA, a chaperone for the H3.3 variant. In addition, both HIRA and H3.3 appear to act cooperatively with the Polycomb group of epigenetic repressors. These results support the involvement of H3.3-mediated nucleosome turnover in restoring the repressed state. The study proposes a regulatory framework integrating transcriptional pausing, histone modification, nucleosome architecture and turnover for cell lineage maintenance. |
Papadopoulos, D. K., Krmpot, A. J., Nikolic, S. N., Krautz, R., Terenius, L., Tomancak, P., Rigler, R., Gehring, W. J. and Vukojevic, V. (2015). Probing the kinetic landscape of Hox transcription factor-DNA binding in live cells by massively parallel Fluorescence Correlation Spectroscopy. Mech Dev 138 Pt 2: 218-225. PubMed ID: 26428533
Summary: Hox genes encode transcription factors that control the formation of body structures, segment-specifically along the anterior-posterior axis of metazoans. Hox transcription factors bind nuclear DNA pervasively and regulate a plethora of target genes, deploying various molecular mechanisms that depend on the developmental and cellular context. To analyze quantitatively the dynamics of their DNA-binding behavior, confocal laser scanning microscopy (CLSM), single-point fluorescence correlation spectroscopy (FCS), fluorescence cross-correlation spectroscopy (FCCS) and bimolecular fluorescence complementation (BiFC) were used. The Hox transcription factor Sex combs reduced (Scr) was shown to form dimers that strongly associate with its specific fork head binding site (fkh250) in live salivary gland cell nuclei. In contrast, dimers of a constitutively inactive, phospho-mimicking variant of Scr show weak, non-specific DNA-binding. These studies reveal that nuclear dynamics of Scr is complex, exhibiting a changing landscape of interactions that is difficult to characterize by probing one point at a time. Therefore, mechanistic evidence is provided using massively parallel FCS (mpFCS). Scr dimers were found to predominantly formed on the DNA and are equally abundant at the chromosomes and an introduced multimeric fkh250 binding-site, indicating different mobilities, presumably reflecting transient binding with different affinities on the DNA. These proof-of-principle results emphasize the advantages of mpFCS for quantitative characterization of fast dynamic processes in live cells. |
Eksi, S. E., Barmina, O., McCallough, C. L., Kopp, A. and Orenic, T. V. (2018). A Distalless-responsive enhancer of the Hox gene Sex combs reduced is required for segment- and sex-specific sensory organ development in Drosophila. PLoS Genet 14(4): e1007320. PubMed ID: 29634724
Summary: Hox genes are involved in the patterning of animal body parts at multiple levels of regulatory hierarchies. Early expression of Hox genes in different domains along the embryonic anterior-posterior (A/P) axis in insects, vertebrates, and other animals establishes segmental or regional identity. However, Hox gene function is also required later in development for the patterning and morphogenesis of limbs and other organs. In Drosophila, spatiotemporal modulation of Sex combs reduced (Scr) expression within the first thoracic (T1) leg underlies the generation of segment- and sex-specific sense organ patterns. High Scr expression in defined domains of the T1 leg is required for the development of T1-specific transverse bristle rows in both sexes and sex combs in males, implying that the patterning of segment-specific sense organs involves incorporation of Scr into the leg development and sex determination gene networks. This study sought to gain insight into this process by identifying the cis-and trans-regulatory factors that direct Scr expression during leg development. Two cis-regulatory elements were identified that control spatially modulated Scr expression within T1 legs. One of these enhancers directs sexually dimorphic expression and is required for the formation of T1-specific bristle patterns. The Distalless and Engrailed homeodomain transcription factors act through sequences in this enhancer to establish elevated Scr expression in spatially defined domains. This enhancer functions to integrate Scr into the intrasegmental gene regulatory network, such that Scr serves as a link between leg patterning, sex determination, and sensory organ development. |
Maiti, S., Acharya, B., Boorla, V. S., Manna, B., Ghosh, A. and De, S. (2019). Dynamic studies on intrinsically disordered regions of two paralogous transcription factors reveal rigid segments with important biological functions. J Mol Biol. PubMed ID: 30802457
Summary: Long stretches of intrinsically disordered regions (IDRs) are abundantly present in eukaryotic transcription factors. Although their biological significance is well appreciated, the underlying structural and dynamic mechanisms of their function are still not clear. Using solution NMR spectroscopy, the structural and dynamic features of two paralogous HOX transcription factors, SCR and DFD, from Drosophila were studied. Both proteins have a conserved DNA-binding homeodomain and a long stretch of functionally important IDR. Using NMR dynamics, flexibility of each residue in these proteins was determined. The flexibility of the residues in the disordered region is not uniform. In both proteins, the IDRs have short stretches of consecutive residues with relatively less flexibility, that is, higher rigidity. One such rigid segment is specifically recognized by another co-transcription factor, thus highlighting the importance of these rigid segments in IDR-mediated protein-protein interactions. Using molecular dynamics simulation, it was further showm that the rigid segments sample less conformations compared to the rest of the residues in the disordered region. The restrained conformational sampling of these rigid residues should lower the loss in conformational entropy during their interactions with binding partners resulting in sequence specific binding. This work provides experimental evidence of a "rigid-segment" model of IDRs, where functionally important rigid segments are connected by highly flexible linkers. Furthermore, a comparative study of IDRs in paralogous proteins reveals that in spite of low-sequence conservation, the rigid and flexible segments are sequentially maintained to preserve related functions and regulations of these proteins. |
Banerjee, A. and Percival-Smith, A. (2020). Post-translational modifications of Drosophila melanogaster HOX protein, Sex combs reduced. PLoS One 15(1): e0227642. PubMed ID: 31931520
Summary: Homeotic selector (HOX) transcription factors (TFs) regulate gene expression that determines the identity of Drosophila segments along the anterior-posterior (A-P) axis. The current challenge with HOX proteins is understanding how they achieve their functional specificity while sharing a highly conserved homeodomain (HD) that recognize the same DNA binding sites. One mechanism proposed to regulate HOX activity is differential post-translational modification (PTM). As a first step in investigating this hypothesis, the sites of PTM on a Sex combs reduced protein fused to a triple tag (SCRTT) extracted from developing embryos were identified by Tandem Mass Spectrometry (MS/MS). The PTMs identified include phosphorylation at S185, S201, T315, S316, T317 and T324, acetylation at K218, S223, S227, K309, K434 and K439, formylation at K218, K309, K325, K341, K369, K434 and K439, methylation at S19, S166, K168 and T364, carboxylation at D108, K298, W307, K309, E323, K325 and K369, and hydroxylation at P22, Y87, P107, D108, D111, P269, P306, R310, N321, K325, Y334, R366, P392 and Y398. Of the 44 modifications, 18 map to functionally important regions of SCR. Besides a highly conserved DNA-binding HD, HOX proteins also have functionally important, evolutionarily conserved small motifs, which may be Short Linear Motifs (SLiMs). SLiMs are proposed to be preferential sites of phosphorylation. Although 6 of 7 phosphosites map to regions of predicted SLiMs, no support was found for the hypothesis that the individual S, T and Y residues of predicted SLiMs are phosphorylated more frequently than S, T and Y residues outside of predicted SLiMs. |
Bu, S., Lau, S. S. Y., Yong, W. L., Zhang, H., Thiagarajan, S., Bashirullah, A. and Yu, F. (2023). Polycomb group genes are required for neuronal pruning in Drosophila. BMC Biol 21(1): 33. PubMed ID: 36793038
Summary: Pruning that selectively eliminates unnecessary or incorrect neurites is required for proper wiring of the mature nervous system. During Drosophila metamorphosis, dendritic arbourization sensory neurons (ddaCs) and mushroom body (MB) γ neurons can selectively prune their larval dendrites and/or axons in response to the steroid hormone ecdysone. An ecdysone-induced transcriptional cascade plays a key role in initiating neuronal pruning. However, how downstream components of ecdysone signalling are induced remains not entirely understood. This study identified that Scm, a component of Polycomb group (PcG) complexes, is required for dendrite pruning of ddaC neurons. Two PcG complexes, PRC1 and PRC2, are important for dendrite pruning. Interestingly, depletion of PRC1 strongly enhances ectopic expression of Abdominal B (Abd-B) and Sex combs reduced, whereas loss of PRC2 causes mild upregulation of Ultrabithorax and Abdominal A in ddaC neurons. Among these Hox genes, overexpression of Abd-B causes the most severe pruning defects, suggesting its dominant effect. Knockdown of the core PRC1 component Polyhomeotic (Ph) or Abd-B overexpression selectively downregulates Mical expression, thereby inhibiting ecdysone signalling. Finally, Ph is also required for axon pruning and Abd-B silencing in MB γ neurons, indicating a conserved function of PRC1 in two types of pruning. This study demonstrates important roles of PcG and Hox genes in regulating ecdysone signalling and neuronal pruning in Drosophila. Moreover, our findings suggest a non-canonical and PRC2-independent role of PRC1 in Hox gene silencing during neuronal pruning. |
Jiang, Y., Chiu, T. P., Mitra, R., Rohs, R. (2024). Probing the role of the protonation state of a minor groove-linker histidine in Exd-Hox-DNA binding. Biophys J, 123(2):248-259 PubMed ID: 38130056
Summary: DNA recognition and targeting by transcription factors (TFs) through specific binding are fundamental in biological processes. Furthermore, the histidine protonation state at the TF-DNA binding interface can significantly influence the binding mechanism of TF-DNA complexes. Nevertheless, the role of histidine in TF-DNA complexes remains underexplored. This study employed all-atom molecular dynamics simulations using AlphaFold2-modeled complexes based on previously solved co-crystal structures to probe the role of the His-12 residue in the Extradenticle (Exd)-Sex combs reduced (Scr)-DNA complex when binding to Scr and Ultrabithorax (Ubx) target sites. These results demonstrate that the protonation state of histidine notably affected the DNA minor-groove width profile and binding free energy. Examining flanking sequences of various binding affinities derived from SELEX-seq experiments, the relationship between binding affinity and specificity was studied. How histidine protonation leads to increased binding affinity but can lower specificity was uncovered. These findings provide new mechanistic insights into the role of histidine in modulating TF-DNA binding. |
Lei, C., Chen, Z., Hao, Y., Huang, W., Chu, T., Xiao, K., Zhang, C., Zhou, W., Li, C., Chen, X. (2025). Quantitative and site-specific chemoproteomic profiling of O-GlcNAcylation in Drosophila. Bioorg Med Chem, 124:118191 PubMed ID: 40245499
Summary: Protein O-GlcNAcylation plays a crucial role in Drosophila melanogaster development. Dysregulation of O-GlcNAc transferase (sxc/Ogt) and GlcNAcase (Oga) disrupts early embryogenesis and locomotor behavior. It is therefore of great interest to identify and quantitatively analyze O-GlcNAcylation sites in Drosophila. This study performed quantitative and site-specific profiling of O-GlcNAcylation in Drosophila by employing a chemoenzymatic labeling strategy. A total of 2196 unambiguous O-GlcNAcylation sites and 1308 O-GlcNAcylated proteins are identified. Quantitative analysis of O-GlcNAcylation in the head of Drosophila with sxc/Ogt knockdown in GABAergic neurons reveals a reduction in O-GlcNAcylation of several proteins involved in muscle development, consistent with the phenotypic defects observed in sxc/Ogt. RNAi Drosophila. Furthermore, quantitative analysis of O-GlcNAcylation under a high-sugar diet reveals altered O-GlcNAcylation of several proteins associated with obesity and neurological diseases, such as Hex-A and Ankyrin 2. This study not only establishes an effective method for large-scale identification of O-GlcNAcylation sites, but also provides a valuable resource for studying O-GlcNAc biology in Drosophila. |
Sex combs reduced is required for labial and first thoracic segment development. Mutants show a tendency to transform labial into maxillary structures. The mutated labial segment appears to be transformed into something closely resembling the more anterior segment, resulting in duplication of maxillary structures. This simplistic, rather linear view of gene action is belied by more than five years research into the regulation of Sex combs reduced. Complex regulation implies a complex function. The biological tools currently employed are as yet too crude to elucidate what must be a high level of interactive complexity. Current understanding is partial and at times contradictory, like the divergent views of the fabled elephant examined by blind men.
From the vertebrate perspective, the labial segment produces what is perhaps the most ornate and alien of the fly's structures, the proboscis. The proboscis protrudes out and downward from the head, a flexible dipstick adapted to gather liquid nourishment. At its working end hang the labella, or labial palps. These furrowed bulbs gather liquid, and move it upward through a series of collecting channels until it reaches the hollow, straw-like labrum. From here, food continues its upward journey into the pharynx by means of muscular action.
Scr is subject to complex regulation. The gene is sandwiched between chromosomal segments that constitute its regulatory region, but even this area of the DNA is complex. It comprises a span of over 70 kb of DNA extending from Antennapedia at one end, sandwiching in the fushi tarazu gene on the way to doing the same with Scr, and continuing to a point beyond the 3' terminus of Deformed . Thus the regulation region of Scr includes the insertion of fushi tarazu, a critically important pair-rule gene.
The existence of pair-rule gene ftz in the center of a regulatory region for a homeotic gene of the Antennapedia complex has implications beyond current understanding. What is ftz doing there, and how is it co-regulated with Scr?
Regulation of Scr requires silencers as well and these are found in a large fragment between ftz and Antp located 40 kb upstream of the Scr promoter (Gorman, 1995).
The regulation of Scr in most tissues is controlled by multiple regulatory elements; the simple view of a proximal promoter and a structural gene proves to be an inadequate explanation. Expression in the prothorax (thoracic segment number one) is controlled by an element in the first intron and an enhancer on the other side of ftz. Expression in the labial lobe requires sequences in the first exon and intron, sequences on the far side of ftz, as well as other sequences, even further away in the direction of Antennapedia. Expression in the salivary gland requires these same regions and expression in the midgut requires sequences found between Scr and ftz. Expression in the CNS requires sequences in the second intron and the 3' end of ftz, as well as other regions. Given this level of complexity, no simple picture is possible (Gindhart, 1995b).
The complexity of Scr regulation, not at all unexpected considering the existence of site specific regulatory elements in such genes as achaete and even-skipped, nevertheless highlights an unbelieveable level of complexity in the evolution of gene regulation. Here are at least three different uses for the Scr gene: labial differentiation, CNS development and mesodermal development, each with independent gene regulation. It will be some time before the details at this level of complexity are understood in terms of the regulation and function of this gene.
Salivary gland formation in the Drosophila embryo is linked to the expression of Sex
combs reduced. When Scr function is missing, salivary glands do not form, and when Scr is
expressed everywhere, salivary glands form in new places. However, not every cell that expresses Scr
is recruited to a salivary gland fate. Along the anterior-posterior axis, the posteriorly expressed proteins
encoded by the teashirt (tsh) and Abdominal-B (Abd-B) genes block Scr activation of salivary gland
genes. Along the dorsal-ventral axis, the secreted signaling molecule encoded by decapentaplegic
prevents activation of salivary gland genes by Scr in dorsal regions of parasegment 2. Five downstream components in the Dpp signaling cascade required to block salivary gland
gene activation have been identified: two known receptors (the type I receptor encoded by the
thick veins gene and the type II receptor encoded by the punt gene); two of the four known
Drosophila members of the Smad family of proteins which transduce signals from the receptors to the
nucleus [Mothers against dpp (Mad) and Medea (Med)], and a large zinc-finger transcription
factor encoded by the schnurri (shn) gene. The expression patterns of d-CrebA and Trachealess were examined in embryos missing zygotic function of schnurri. In embryos homozygous for shn, a dorsal expansion of salivary gland protein expression is observed. The presence of amnioserosa, an extreme dorsal cell type, suggests that embryos lacking zygotic shn function are not ventralized, as are embryos missing maternal and zygotic function of tkv, pt, Mad, or Med or missing zygotic function of dpp. These results reveal how anterior-posterior and
dorsal-ventral patterning information is integrated at the level of organ-specific gene expression (Henderson, 1999).
Scr is normally expressed in all the cells that form the salivary gland. However, as the salivary gland
invaginates, SCR mRNA and protein disappear. Homeotic genes, such as Scr, specify tissue identity by regulating the
expression of downstream target genes. For many homeotic proteins, target gene specificity is achieved by cooperatively
binding DNA with cofactors. Therefore, it is likely that Scr also requires a cofactor(s) to specifically bind to DNA and
regulate salivary gland target gene expression. Two homeodomain-containing proteins encoded by the
extradenticle and homothorax genes are also required for salivary gland formation. exd and hth function at two
levels: (1) exd and hth are required to maintain the expression of Scr in the salivary gland primordia prior to invagination
and (2) exd and hth are required in parallel with Scr to regulate the expression of downstream salivary gland genes. Scr regulates the nuclear localization of Exd in the salivary gland primordia through repression of homothorax expression, linking the regulation of Scr activity to the disappearance of Scr expression in invaginating salivary
glands (Henderson, 2000).
To determine if Exd cooperates with Scr to control salivary gland gene expression, the accumulation
of two early salivary gland proteins, CrebA and Trachealess
(Trh), was examined in embryos lacking exd function. Zygotic loss of exd
function results in a reduction in the number of
salivary gland cells expressing CrebA and Trh, as well
as a decrease in the level of protein made in these cells. This reduced level of salivary gland
protein expression is not as severe as the one seen in Scr mutant
embryos. Unlike SCR, EXD mRNA is
supplied maternally and, thus,
the maternal contribution may partially compensate for the
loss of zygotic function. To test this possibility, the maternal
contribution of exd was removed using the FLP-FRT
system. In embryos lacking
maternal exd function, salivary gland expression of
CrebA and Trh is at wild-type levels. However, salivary gland expression of CrebA and
Trh is completely absent in embryos lacking both the
maternal and the zygotic contributions of exd. Thus, exd is required for embryonic salivary
gland gene expression. Moreover, zygotically provided exd
can rescue the loss of maternally provided exd and maternally
provided exd can partially compensate for zygotic loss
of exd (Henderson, 2000).
Since Scr, exd, and hth are required for salivary gland
formation, the mRNA and/or protein expression
patterns of these genes during normal salivary gland
formation were examined. During stages 9 and 10, when salivary gland gene
expression is established, Scr and hth are expressed in the
salivary gland primordia, as well as other tissues, and Hth
and Exd are nuclear. During stage 11, after the establishment of early
salivary gland gene expression, the salivary glands begin to
invaginate. At this stage, there are several changes in the
expression and/or localization of these genes and/or proteins
in the salivary gland cells: Scr and hth transcripts
disappear, Hth protein disappears, and Exd protein becomes
cytoplasmic (Henderson, 2000).
Hth is known to regulate the nuclear entry of Exd in
other cells of the embryo and in the imaginal discs. In
embryos lacking hth function, Exd is cytoplasmic throughout
the embryo, including the cells of the salivary gland
primordia. Since Hth is required for the activity of Exd,
and Exd is required to maintain Scr expression in the
salivary gland primordia, the expression
of Scr in embryos lacking hth function was also examined. As observed in
exd minus embryos, neither SCR mRNA nor Scr protein expression
is maintained in the ventral cells of PS2 in hth mutants. Exd is required for the stability
of Hth throughout the embryo and, correspondingly,
a loss in Hth protein stability, but not HTH transcript
accumulation is observed in exd mutant embryos. Thus,
hth maintains the expression of Scr in the salivary gland
primordia and regulates the nuclear localization of Exd
throughout the embryo, including the cells of the salivary
gland primordia. Furthermore, exd is required for accumulation
of Hth (Henderson, 2000).
Scr, Exd, and Hth are expressed in the salivary gland
primordia when salivary gland gene expression is established.
Once the salivary gland cells begin to invaginate, the
expression and/or localization of all three proteins changes
in the salivary gland primordia. This result indicates that a
gene(s) expressed in the salivary gland primordia during
stage 11 is required for these changes in gene expression and
protein localization. Since hth expression is specifically lost
in the salivary gland primordia, a test was performed to see if Scr regulates hth
expression in the salivary gland primordia. In embryos
lacking Scr function, HTH mRNA and Hth protein are
maintained in the cells that would normally form the
salivary gland beyond stage 11. Correspondingly, Exd remains
nuclear in the ventral cells of PS2 in embryos lacking Scr
function. Therefore Scr is required to
repress hth expression in the cells of the salivary gland
primordia during stage 11. The loss of hth expression
consequently leads to the loss of nuclear Exd accumulation (Henderson, 2000).
Since Scr may act either directly or indirectly to shut off
hth expression, the expression of hth was examined in
embryos mutant for two early salivary gland genes that
encode transcription factors: fkh, which encodes a winged-helix
transcription factor related to mammalian HNF-3beta, and Creb-A, which encodes a beta-Zip
protein that binds cyclic-AMP-response elements. No change in hth expression is seen in
embryos lacking either fkh or Creb-A function. Therefore, Scr does not act through fkh or
Creb-A to shut off hth transcription.
These findings suggest that prior to stage 11, Hth regulates
the nuclear entry of Exd in the salivary gland primordia.
During stage 11, Hth is no longer expressed in these cells
and, as a consequence, Exd localizes to the cytoplasm.
Without Exd and Hth in the nucleus, Scr expression is not
maintained. To test this idea, hth expression
was maintained in cells of the salivary gland primordia using the
GAL4-UAS system. This expression
pattern was achieved using a fkh-GAL4 construct
to drive expression of UAS-hth in the
salivary gland secretory cell primordia from stage 10 onward.
When hth was expressed under the control of the
fkh-GAL4 driver, Hth protein could be detected in the
salivary gland nuclei throughout embryogenesis. Correspondingly, Exd and Scr were
detected in the salivary gland until late stages of embryogenesis. These results
indicate that Hth is not only necessary, but is also sufficient,
to maintain Scr expression in the salivary gland (Henderson, 2000).
In thoracic and abdominal segments of Drosophila, the expression pattern of Bithorax-Complex Hox genes is known to specify the segmental identity of neuroblasts (NB) prior to their delamination from the neuroectoderm. This study identified and characterized a set of serially homologous NB-lineages in the gnathal segments and used one of them (NB6-4 lineage) as a model to investigate the mechanism conferring segment-specific identities to gnathal NBs. It was shown that NB6-4 is primarily determined by the cell-autonomous function of the Hox gene Deformed (Dfd). Interestingly, however, it also requires a non-cell-autonomous function of labial and Antennapedia that are expressed in adjacent anterior or posterior compartments. The secreted molecule Amalgam (Ama) was identified as a downstream target of the Antennapedia-Complex Hox genes labial, Dfd, Sex combs reduced and Antennapedia. In conjunction with its receptor Neurotactin (Nrt) and the effector kinase Abelson tyrosine kinase (Abl), Ama is necessary in parallel to the cell-autonomous Dad pathway for the correct specification of the maxillary identity of NB6-4. Both pathways repress CyclinE (CycE) and loss of function of either of these pathways leads to a partial transformation (40%), whereas simultaneous mutation of both pathways leads to a complete transformation (100%) of NB6-4 segmental identity. Finally, the study provides genetic evidences, that the Ama-Nrt-Abl-pathway regulates CycE expression by altering the function of the Hippo effector Yorkie in embryonic NBs. The disclosure of a non-cell-autonomous influence of Hox genes on neural stem cells provides new insight into the process of segmental patterning in the developing CNS (Becker, 2016).
The Drosophila head consists of seven segments (4 pregnathal and 3 gnathal) all of which contribute neuromeres to the CNS. The brain is formed by approximately 100 NBs per hemisphere, which have been individually identified and assigned to specific pregnathal segments [The anterior pregnathal region (procephalon) is composed of the labral, ocular, antennal, intercalary segments, see Segment polarity and DV patterning gene expression reveals segmental organization of the Drosophila brain]. As judged from comparison of the combinatorial codes of marker gene expression only few brain NBs appear to be serially homologous to NBs in the thoracic/abdominal ventral nerve cord, reflecting the highly derived character of the brain neuromeres. The connecting tissue between brain and the thoracic VNC consists of three neuromeres formed by the gnathal head segments named mandibular (mad), maxillary (max) and labial (lab) segment, but the number and identity of the neural stem cells and their lineage composition in these segments is still unknown. Compared to the thoracic ground state the segmental sets of gnathal NBs might be reduced to different degrees, but are thought to be less derived compared to the brain NBs. Therefore, to fully understand segmental specification during central nervous system development, it is important to identify the neuroblasts and their lineages in these interconnecting segments (Becker, 2016).
Assuming that most NBs in the gnathal segments still share similarities to thoracic and abdominal NBs, this study sought serially homologous NB-lineages, which are suitable for genetic analyses. Using the molecular marker eagle (eg), which specifically labels four NB-lineages in thoracic/abdominal hemisegments this study identified three serial homologs (NB3-3, NB6-4 and NB7-3) in the gnathal region. To investigate the mechanisms conferring segmental identities, focus was placed on one of them, the NB6-4 lineage, which shows the most significant segment-specific modifications. The analysis reveals a primary role of the Antennapedia-Complex (Antp-C) Hox gene Deformed (Dfd) in cell-autonomously specifying the maxillary fate of NB6-4 (NB6-4max). Surprisingly, an additional, non-cell-autonomous function was uncovered of the Antp-C Hox genes labial (lab, expressed anterior to Dfd) and Antennapedia (Antp, expressed posterior to Dfd) in specifying NB6-4max. In a mini-screen for downstream effectors the secreted protein Amalgam (Ama) was identified as being positively regulated by lab, Dfd and Antp and negatively regulated by the Antp-C Hox gene Sex combs reduced (Scr). Loss of function of Ama and its receptor Neurotactin (Nrt) as well as the downstream effector kinase Abelson tyrosine kinase (Abl) lead to a transformation of NB6-4max similar to Dfd single mutants. Thus, in parallel to the cell-autonomous role of Dfd, a non-cell-autonomous function of Hox genes lab and Antp, mediated via the Ama-Nrt-Abl pathway, is necessary to specify NB6-4max identity. Disruption of either of these pathways leads to a partial misspecification of NB6-4max (approx. 40%), whereas simultaneous disruption of both pathways leads to a complete transformation (approx. 100%) of NB6-4max to a labial/thoracic identity. It was further shown that both pathways regulate the expression of the cell cycle gene CyclinE, which is necessary and sufficient to generate labial/thoracic NB6-4 identity. Whereas Dfd seems to directly repress CyclinE transcription (similar to AbdA/AbdB in the trunk), indications are provided that the Ama-Nrt-Abl pathway prevents CyclinE expression by altering the activity of the Hippo/Salvador/Warts pathway effector Yorkie (Yki) (Becker, 2016).
Along the anterior-posterior axis the CNS consists of segmental units (neuromeres) the composition of which is adapted to the functional requirements of the respective body parts. In Drosophila the CNS comprises 10 abdominal, three thoracic, three gnathal and four pregnathal (brain) neuromeres that are generated by stereotyped populations of neural stem cells (neuroblasts, NBs). The pattern of NBs in thoracic segments resembles the ground state while NB patterns in the other segments are derived to various degrees. Within each segment individual NBs are specified by positional information in the neuroectoderm. NBs delaminating from corresponding positions in different segments express similar sets of molecular markers, generate similar lineages, and are called serial homologs. However, for thoracic and abdominal neuromeres it has been shown that the composition of a number of serially homologous NB-lineages shows segment-specific differences. In the more derived gnathal and pregnathal head segments embryonic NB-lineages and the mechanisms of their segmental specification have not been analyzed so far (Becker, 2016).
Using the well-established molecular marker Eagle (Eg) which labels four embryonic NB-lineages (NB2-4, NB3-3, NB6-4, NB7-3) in all thoracic and most of the abdominal segments this study identified serially homologous lineages of NB3-3, NB6-4 and NB7-3 in gnathal segments. The embryonic NB7-3 lineage shows segmental differences as it comprises increasing cell numbers from mandibular (2 cells), maxillary (3 cells) to labial (3-5 cells) segments, while cell numbers are decreasing from T1-T2 (4 cells), T3-A7 (3 cells) to A8 (2-3 cells). Reduced cell numbers in the mandibular and maxillary NB7-3 lineages depend on Dfd and Scr function, respectively . While NB7-3 appeared in all three gnathal segments, NB3-3 and NB6-4 was only found in labial and maxillary segments, and NB2-4 was not found in any of them. Preliminary data suggest that the missing NBs are not generated in these segments, instead of being eliminated by apoptosis. For the terminal abdominal neuromeres (A9, A10) it has recently been shown that the formation of a set of NBs (including NB7-3) is inhibited by the Hox gene Abdominal-B. Similarly, in Dfd mutants the formation was observed of a NB with NB6-4 characteristics in mandibular segments (10%), in which it is never found in wild type (Becker, 2016).
Similar to the thoracic and abdominal segments NB6-4 showed dramatic differences between maxillary and labial segments. NB6-4max produces glial cells only (like abdominal NB6-4), whereas the labial homolog produces neurons in addition to glial cells (like thoracic NB6-4). The number of glial cells produced by the glioblasts NB6-4max (4 cells) and abdominal NB6-4 (2 cells) and by the neuroglioblasts NB6-4lab (3 glia) and thoracic NB6-4 (3 glia) is segment-specific(Becker, 2016).
Thus segment-specific differences among serially homologous lineages may concern types and/or numbers of specific progeny cells and may result from differential specification of NBs and their progeny, differential proliferation and/or differential cell death of particular progeny cells. It has been shown that the segment-specific modification of serially homologous lineages is under the control of Hox genes and that during neurogenesis Hox genes act on different levels, i.e. they act in a context-specific manner at different developmental stages and in different cells. In the thoracic/abdominal region segmental identity is conferred to NBs early in the neuroectoderm by cell-autonomous function of Hox genes of the Bithorax-Complex. This study used the NB6-4 lineage to clarify mechanisms of segmental specification in the gnathal segments (Becker, 2016).
In segments of the trunk, the action of Hox genes strictly follows the rule of the posterior prevalence concept: More posterior expressed Hox genes repress anterior Hox genes and thereby determine the segmental identities. In the gnathal segments this phenomenon was not observed on the level of the nervous system. Removing Hox genes of the Antp-C had no or only minor impact on the expression domain of other Antp-C Hox genes. Similar results were also obtained in a study that analyzed cross-regulation of Hox genes upon ectopic expression (Becker, 2016).
Moreover, it seems that at least in the case of the differences monitored between labial and maxillary segments Hox gene function has to be added to realize the more anterior fate. Antennapedia has no impact on NB6-4 identity in the labial segment, but specification of the maxillary NB6-4 requires the function of Deformed and Sex combs reduced. These two Hox genes are not repressed or activated by Antp. Also, cross-regulation between Dfd and Scr seems to be unlikely or is very weak since only mild effects were observed on the protein level and on the phenotypic penetrance. In principle Scr can repress Dfd, but it was suggested that this occurs only when products are in sufficient amounts. In NB6-4 Dfd and Scr are co-expressed, but Scr levels appear to be insufficient to repress Dfd. Dfd seems to be the major Hox gene that cell-autonomously confers the maxillary NB6-4 fate, since the loss of Dfd showed the highest transformation rate and, more importantly, ectopic expression of Dfd in thoracic segments leads to a robust transformation towards maxillary fate. Scr does not act redundantly since in double mutants Dfd/Scr no synergistic effect was observed. It might have a fine-tuning effect, as it was shown that Scr influences Ama by repressing its transcription, whereas all other Antp-C Hox genes seem to activate Ama. However, since only minor changes were found in cell identities and numbers in Scr LoF background, the role of Scr in NB6-4max stays enigmatic (Becker, 2016).
Surprisingly cell-autonomous Hox gene function was not the only mechanism that confers segmental identity in NB6-4max. Loss of Dfd showed an effect in approx. 43% of all segments. Moreover, mutations of the adjacently expressed Hox genes labial and Antennapedia in combination with Dfd LoF showed a dramatic increase in the transformation rate of NB6-4max. Their expression patterns on the mRNA and protein level were carefully studied in wild type and Hox mutant background. In no case were these genes found to be expressed in NB6-4max or in the neuroectodermal region from which NB6-4max delaminates. This indicates that labial and Antennapedia influence NB6-4max fate in a non-cell-autonomous manner. That Hox genes can act non-cell-autonomously on stem cells was recently shown in the male germ-line, were AbdB influences centrosome orientation and the proliferation rate through regulation of the ligand Boss in the Sevenless-pathway. In this study Antp-C Hox genes controled the expression of the secreted molecule Amalgam, which spreads to adjacent segments and ensures segmental specification of NB6-4max in a parallel mechanism to the cell-autonomous function of Dfd. Thus, this study provides first evidence for parallel non-cell-autonomous and cell-autonomous functions of Antp-C genes during neural stem cell specification in the developing CNS (Becker, 2016).
Abelson kinase (Abl) was shown to be required for proper development of the Drosophila embryonic nervous system. In neurons Abl interacts with proteins like Robo or Chickadee and influences the actin cytoskeleton in the growth cone to regulate axonogenesis and pathfinding. In this system it was also demonstrated that Ama and Nrt are dominant modifiers of the Abl phenotype. It is proposed that the interaction of secreted Ama and the membrane-bound Nrt regulates Abl function in NBs. This leads to the correct segmental specification of NB6-4max. Antp-C Hox genes lab, Antp and Dfd regulate the expression of Ama and in mutants for theses Hox genes expression of Ama is severely reduced, which leads to the transformation of NB6-4max due to missing Abl function and de-repression of the cell cycle gene CyclinE. That Abl can influence the expression of CyclinE was also demonstrated in a modifier-screen in the Drosophila eye, but the mechanism remained unclear. Genetic analysis now suggests that in NBs this might occur via the regulation of the highly conserved Hippo-Salvador-Warts pathway and its downstream transcriptional co-activator Yki, which is known to regulate CyclinE expression. The Hippo-Salvador-Warts pathway controls organ growth and cell proliferation in Drosophila and vertebrates but so far has not been implicated in embryonic NB development. This study observed Yki cytoplasmic localization in wild type NB6-4max prior to division suggesting the active Hippo pathway. Nuclear localization of Yki could not be detected in Abl mutants, the loss of Yki activity in the Abl mutant background leads to a significant reduction in the strength of the Abl single mutant phenotype showing their genetic interaction and therefore supporting the proposed model in which Abl influences Yki activity. Moreover, expression of constitutive active Yki also lead to the transformation of NB6-4max and phenotypes that were similar to those observed in Abl mutants. Attempts were made to assess how Abl might influence Yki activity. Work in vertebrates suggests that this could be at least on two levels: first, c-Abl was shown to directly phosphorylate and activate the vertebrate MST1 and MST2 (Hpo homologue) and the Drosophila Hpo on a conserved residue (Y81) and second, c-Abl can also phosphorylate YAP1, which changes its function to become pro-apoptotic. This analysis suggests that in NBs Abl might regulate Hpo, since changes were found in the stability of Salvador, which is used as a Hpo activity readout, but a parallel direct regulation of Yki could not be ruled out, since it was recently shown that other pathways like the AMPK/LKB1 pathway can directly influence Yki activity. Since severe over-proliferation was observed in Abl or lab/Dfd mutants, that have an impaired Ama-Nrt-Abl pathway, or upon overexpression of YkiCA, future studies need to elucidate whether and how the proto-oncogene Abl kinase and Hox genes act on growth and proliferation or even tumor initiation through regulation of the Hippo/Salvador/Warts pathway (Becker, 2016).
Formation of self-associating loop domains is a fundamental organizational feature of metazoan genomes. This study employed quantitative live-imaging methods to visualize impacts of higher-order chromosome topology on enhancer-promoter communication in developing Drosophila embryos. Evidence is provided that distal enhancers effectively produce transcriptional bursting from target promoters over distances when they are flanked with boundary elements. Importantly, neither inversion nor deletion of a boundary element abrogates this 'enhancer-assisting activity,' suggesting that they can facilitate intra-domain enhancer-promoter interaction and production of transcriptional bursting independently of topologically associating domain (TAD) formation. In contrast, domain-skipping activity of distal enhancers was lost after disruption of topological domains. This observation raises a possibility that intra-domain and inter-domain enhancer-promoter interactions are differentially regulated by chromosome topology (Yokoshi, 2020).
This study presents evidence that boundary elements help distal enhancers to induce transcriptional bursting from their target promoters. The data suggest that TAD formation is not a prerequisite for facilitating intra-domain enhancer-promoter interaction because inversion or deletion of Homie did not abrogate this 'enhancer-assisting activity.' Supporting this view, deletion of SF1 boundary element from the endogenous locus did not eliminate ftz stripe formation. In contrast, analysis of the neighboring Hox gene Scr revealed that domain-skipping activity of distal T1 enhancer relies on pairing between SF1 and SF2 boundary elements, suggesting that intra-domain and inter-domain enhancer-promoter interactions have differential dependence on TAD formation (Yokoshi, 2020).
It is well established that TAD formation is mainly regulated by a Zn-finger DNA-binding protein CTCF and the ring-shaped SMC complex cohesin in vertebrates. When CTCF or Rad21, a kleisin subunit of cohesin, is acutely removed by auxin-inducible degron, essentially all TADs are lost in mammalian cultured cells. Consistent with this, knockout of cohesin-loading factor Nipbl leads to loss of TADs in mouse liver. Importantly, these studies agree well in a point that elimination of TADs has a modest influence on overall transcription activities. Thus, topological boundaries and resulting TAD structures have been implicated to play an auxiliary role in enhancer-promoter communication and gene expression. This study combined quantitative live imaging and genome-editing methods to directly visualize impacts of higher-order genome topology on enhancer-promoter interaction. The data suggest that TAD formation itself has minor influence on intra-domain enhancer-promoter interaction and spatial patterning of gene expression because only ~20% reduction was seen in the total output after Homie inversion (Nhomie/Homie versus Nhomie/Homieinv) and ~10%-13% reduction after SF1 deletion. Consistent with this, recent whole-genome sequencing study of balancer chromosomes that contain massive rearrangements of genome topology revealed that alternation of TAD structures has minor impacts on overall level and specificity of gene expression in Drosophila. Similarly, in mammalian system, genome-editing analysis of Sox9 locus showed that TAD disruption results in only ~10%-15% reduction of Sox9 expression and no detectable phenotypes in developing mouse limb buds, again suggesting non-essential role of TADs in gene expression. Supporting this view, it was recently reported that Drosophila CTCF-null mutant can progress through embryogenesis. Moreover, it is known that C. elegans and Arabidopsis lack CTCF and TAD-like structures, yet they achieve productive gene expression during development. It is speculated that TAD-mediated intra-domain interactions confer robustness of transcriptional program under environmental fluctuations, such as temperature change and nutrient availability, thereby providing selective advantages in a population. Contrary to intra-domain regulatory interactions, the data clearly showed that inter-domain T1-Scr interaction was compromised in ΔSF1 embryos, suggesting that pairing of boundary elements is required for gene-skipping activity of distal enhancers. Given an essential role of SF1 in correct Scr expression, it might be possible that ftz TAD is formed as a consequence of bringing distal T1 enhancer and Scr promoter together by looping out an intervening ftz transcription unit. Importantly, ΔSF1 mutant embryos developed normally even without early Scr expression, suggesting that enhancer redundancy acts as an alternative mechanism to achieve reliable gene expression under various genetic perturbations, including misregulation of TAD formation (Yokoshi, 2020).
The data suggest that boundary elements are capable of facilitating enhancer-promoter communication independently of TAD formation because the level of gene activities remained to be substantially higher even after Homie inversion or deletion. As in mammalian genome, TAD boundaries are enriched for CTCF and Rad21 in Drosophila. In past studies, much effort has been focused on functional analysis of these proteins because CTCF and cohesin have been long known to play a fundamentally important role in genome organization. However, these are not the only proteins enriched at boundaries. Whole-genome ChIP assays revealed that so-called insulator proteins, such as Su(Hw), Mod(mdg4), CP190, and BEAF-32, colocalize with CTCF-binding sites in the Drosophila genome. Moreover, other regulatory proteins, such as Fs(1)h, DREF, Chromator, L3mbt, Z4 (also known as Putzig), TFIIIC, and condensin subunits (CAP-H2 and Barren), are shown to be a major constituent of the complex at TAD boundaries. Indeed, Nhomie and Homie used in this study are highly enriched for these components. Among these, Fs(1)h is known as a Drosophila homolog of bromodomain-containing protein Brd4. Recent studies suggested that Brd4 undergoes dynamic liquid-liquid phase separation through the conserved intrinsically disordered region at C-terminal domain, producing transcriptional condensates or transcription hubs that colocalize with the Mediator and Pol II clusters. In addition, Brd4 is known to interact with the positive transcription elongation factor P-TEFb, a complex of Cdk9 and its regulatory subunit cyclin T1. Intriguingly, P-TEFb is also suggested to undergo liquid-liquid phase separation though the conserved histidine-rich domain of cyclin T1 to increase processivity of Ser2 phosphorylation at Pol II CTD. Thus, it might be possible that boundary-enriched Fs(1)h acts as a scaffold to increase local concentration of transcription factors, co-activators, elongation factors, and Pol II complexes, thereby affecting the timing and the size of transcriptional bursting independently of TAD formation. Importantly, other boundary-enriched proteins DREF, Chromator, and Z4 are also suggested to be involved in recruitment of Pol II into a subset of genes, giving rise to a possibility that these proteins further contribute to assembly of transcription hubs at nearby enhancers. It appears that this mechanism is particularly important for distal enhancers to overcome large genomic separation because extension of enhancer-promoter distance from 6.5 kb to 9 kb leads to significant (more than 50%) diminishment in the level of total output when boundary elements are not present. It is speculated that this is a common regulatory mechanism in the Drosophila genome because typical enhancer-promoter separation is estimated to be ~10 kb. Future functional studies should elucidate the molecular mechanism underlying 'enhancer-assisting activity' of boundary elements (Yokoshi, 2020).
Feng, S., Rastogi, C., Loker, R., Glassford, W. J., Tomas Rube, H., Bussemaker, H. J. and Mann, R. S. (2022). Transcription factor paralogs orchestrate alternative gene regulatory networks by context-dependent cooperation with multiple cofactors. Nat Commun 13(1): 3808. PubMed ID: 35778382
In eukaryotes, members of transcription factor families often exhibit similar DNA binding properties in vitro, yet orchestrate paralog-specific gene regulatory networks in vivo. The serially homologous first (T1) and third (T3) thoracic legs of Drosophila, which are specified by the Hox proteins Scr and Ubx, respectively, offer a unique opportunity to address this paradox in vivo. Genome-wide analyses using epitope-tagged alleles of both Hox loci in the T1 and T3 leg imaginal discs, the precursors to the adult legs and ventral body regions, show that ~8% of Hox binding is paralog-specific. Binding specificity is mediated by interactions with distinct cofactors in different domains: the Hox cofactor Exd acts in the proximal domain and is necessary for Scr to bind many of its paralog-specific targets, while in the distal leg domain, the homeodomain protein Distal-less (Dll) enhances Scr binding to a different subset of loci. These findings reveal how Hox paralogs, and perhaps paralogs of other transcription factor families, orchestrate alternative downstream gene regulatory networks with the help of multiple, context-specific cofactors (Feng, 2022).
This study used a combination of whole-genome and mechanistic approaches to understand how serially homologous appendages, such as the fly T1 and T3 legs, obtain their unique morphologies due to the activities of parallel Hox gene networks. The very similar transcriptomes in the three pairs of leg discs suggest that the different morphologies are largely a consequence of changing the expression patterns of the same sets of genes. By comparing the genome-wide DNA-binding profiles of the two relevant Hox paralogs, Scr and Ubx, in their native physiological contexts, this study found hundreds of paralog-specific Hox targets, accounting for ~8% of all binding events for these two Hox proteins. Next, differences in chromatin accessibility and Hox monomer binding preferences were shown to be unlikely to account for paralog-specific binding. Instead, it was demonstrated that interaction with the Hox cofactor Exd explains a large fraction of Scr's paralog-specific binding events. Finally, this study identified Dll as a Hox cofactor in the complementary distal domain of the leg disc. Results from RNA-seq, CBP ChIP, and reporter assays suggest that about 1/3 of the paralog-specific Scr-binding events are functional and lead to tissue-specific gene regulation. Thus, paralog-specific Hox-DNA binding, which is mediated by multiple cofactors including Exd and Dll, contribute significantly to paralog-specific Hox gene networks (Feng, 2022).
Previous in vitro studies provided compelling evidence that the DNA-binding specificities of different Hox-Exd dimers are more divergent from each other than those of Hox monomers, a phenomenon termed latent specificity. There have also been several in vivo examples in which paralog-specific Hox-DNA binding and target regulation was shown to depend on an interaction with Exd. This study shows that, on a genome-wide scale, the interaction with Exd explains a significant fraction of paralog-specific Hox binding, which often leads to paralog-specific gene regulation (Feng, 2022).
Earlier work also suggested that there is a tradeoff between specificity and affinity for Hox-Exd-binding motifs, where high affinity binding motifs are more likely to have low specificity for different Hox-Exd heterodimers. The paralog-specific, Exd-dependent CRMs characterized in this study (ac-1, h-1, and fj<-1), have higher affinity Hox-Exd-binding motifs than those previously described in the shavenbaby (svb) gene: the major Scr-Exd motif in the fj-1 CRM has an affinity of about 0.06 relative to the optimal motif in the genome, while the motifs in ac-1-1 and h-1 have even higher relative affinities of nearly 0.15 and 0.2, respectively. In contrast, the Ubx-Exd-binding motifs in CRMs from svb have a relative affinity of <0.0118. One possible explanation for this difference is that the svb CRMs are active in embryos, which have many different cell types, while the CRMs characterized in this study are active in leg discs, which have significantly less cell-type complexity. Embryonic CRMs may require especially low-affinity binding motifs to distinguish their activities in a context with many cell types. Consistent with this idea, the fkh250 CRM, which is also active in embryos, uses an Scr-Exd-binding motif with a low relative affinity of 0.01718,36. Notably, the relative affinities for the Scr-Exd-binding motifs in ac-1, h-1, and fj-1 are at least eightfold higher than for Ubx-Exd. Manual inspection of other intergenic and intronic loci with ScrT1 > UbxT3 binding suggests that there are many other CRMs that follow this same rule. Thus, for specificity to occur, the most relevant feature may be that the affinity for the 'correct' TFs, in this case Scr-Exd, must be significantly greater compared to the affinity for other 'incorrect' TFs that are co-expressed in the same or homologous cells (Feng, 2022).
Because Exd is only nuclear in a subset of cells during Drosophila development, such as the proximal domain of the leg disc, it was unlikely that Exd was the only Hox cofactor. In fact, CRMs that are directly regulated by Ubx have been described in cells where Exd is not available to be a cofactor. However, it has remained an unresolved question whether non-Exd cofactors are used in these examples. More generally for the leg imaginal disc, the entire distal domain, extending from the trochanter to the tarsus, is without nuclear Exd, yet has Hox-dependent segment-specific morphological characteristics, such as the sex combs on the male T1 leg. Although several candidate TFs have been proposed to be Hox cofactors, none have been confirmed. This study provides evidence that Dll is a distally acting Hox cofactor in leg discs (Feng, 2022).
There are many differences between how Exd and Dll interact with Hox proteins when bound to DNA. The Scr-Exd-binding motif is comprised of two partially overlapping half-sites, while the Scr-Dll motif consists of two HD-binding motifs separated by a spacer of several base pairs. Another difference is that the amount of cooperativity observed for Hox-Exd is far greater than that observed for Hox-Dll. The overlapping nature of the Hox- and Exd-binding motifs may be important for latent specificity, which for Scr requires an Exd-induced conformational change of the homeodomain. In contrast, there is no evidence that latent specificity occurs as a consequence of Hox-Dll binding. Instead, the modest cooperativity observed for the Scr-Dll heterodimer is likely a consequence of increasing Scr-DNA-binding affinity via a protein-protein interaction and closely spaced Dll and Scr-binding motifs (Feng, 2022).
More generally, it is suggested that mode of DNA binding exhibited by Hox-Exd, which is highly cooperative and reveals latent specificity, may be the exception rather than the rule for TF-TF interactions within CRMs, and that the Scr-Dll example, with weak cooperativity between TFs stemming from a protein-protein interaction, may be the more common mode of interaction to distinguish the binding of paralogous TFs. In support of this notion, a systematic in vitro study identified 315 TF-TF interactions, only five of which exhibited latent specificity (Feng, 2022).
The TALE homeodomain proteins, which include Exd and Hth, are very ancient TFs that were present before the split of plants and animals, and TALE-mediated nuclear localization analogous to the Hth-Exd example in flies has been described in plants. In contrast, the Hox gene family is only present in metazoans, and Dll is specific to bilaterians. Moreover, it has been proposed that Dll initially functioned in the CNS, and was later co-opted to pattern the distal appendage. Based on these observations, it is plausible that the Hox-Dll interaction evolved more recently than the Hox-Exd interaction, accounting for why Exd interacts with all Hox paralogs, while Dll may be a more limited Hox cofactor. This is supported by the results from a small-scale bimolecular fluorescence complementation (BiFC) screen that revealed Dll interacts with some Hox proteins, but not others (Feng, 2022).
Notably, the combined activities of Exd and Dll still do not account for all ScrT1 > UbxT3-binding events genome-wide and reporter analysis suggests the presence of additional, yet to be identified Hox cofactors that have the capacity to promote Scr-specific binding. It is suggested that the Hox-Dll mode of binding uncovered in this study may be representative of additional TFs that also have the ability to promote paralog-specific Hox binding and activity at specific CRMs. Further, it is noted that the differentiation of the T1 and T3 leg fates is a continuous developmental process and that the observations described in this study are limited to the late 3rd instar stage. Nevertheless, it is expected that the principles governing Hox paralog specificity uncovered in this study will likely extend to other developmental stages and tissues. Finally, although this study focused on the role of paralog-specific TF-DNA binding, there may be additional mechanisms that do not depend on differences in DNA binding between paralogous TFs that also contribute to their specific functions (Feng, 2022).
Molecular mapping of Scr breakpoint lesions has defined a segment of greater than 70 kb of DNA necessary for proper Scr gene function. This region is split by the fushi tarazu (ftz) gene, with lesions affecting embryonic Scr function molecularly mapping to the region proximal (5') to ftz and those exhibiting polyphasic semilethality predominantly mapping distal (3') to ftz (Pattatucci, 1991a). Distal to ftz is Antennapedia, 30 kb from ftz, and 48 kb from Scr. Deformed is 20 kb downstream of Scr (Le Motte, 1989).
Genomic length - 23 kb
cDNA clone length - There are two transcripts, 3.6 and 5.2 kb consisting of alternative 3' ends (Andrew, 1995).
Bases in 5' UTR - 626
Exons - three
Bases in 3' UTR - 2261
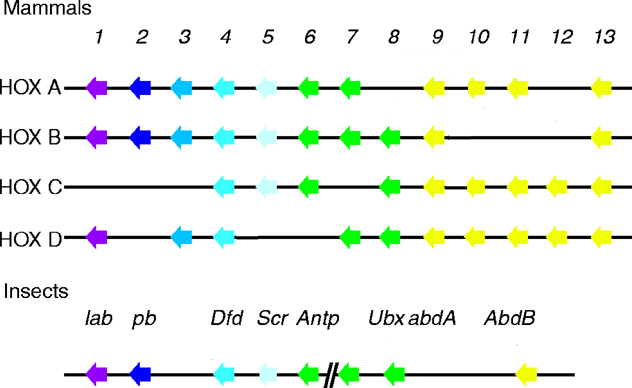
Scr has sequence homology to mouse HOXA5, B5 and C5 (see four paralogous Hox clusters of mammals). The greatest similarity is within the DNA-binding homeodomain. Two shorter conserved motifs are found: one near the N-terminus and a second in a region N-terminal to the homeodomain. The former consists of an MSSF sequence that differs by one amino acid from the MSSY consensus as with most of the D. melanogaster homeotic genes. The latter consists of a WPWM core sequence (LeMotte, 1989 and Andrew, 1995).
date revised: 25 March 2026
Home page: The Interactive Fly © 2026 Thomas B. Brody, Ph.D.
The Interactive Fly resides on the
Society for Developmental Biology's Web server.
{kind=link}