The Interactive Fly
Zygotically transcribed genes
The TOR signaling pathway is a portion of the Growth response - The Insulin receptor signaling pathway
TORC1 (target of rapamycin complex 1) has a crucial role in the regulation of cell growth and size. A wide range of signals, including amino acids, is known to activate TORC1. This study reports the identification of Rag GTPases (RagA-B and RagC-D) as activators of TORC1 in response to amino acid signals. Knockdown of Rag gene expression suppressed the stimulatory effect of amino acids on TORC1 in Drosophila melanogaster S2 cells. Expression of constitutively active (GTP-bound) Rag in mammalian cells activated TORC1 in the absence of amino acids, whereas expression of dominant-negative Rag blocked the stimulatory effects of amino acids on TORC1. Genetic studies in Drosophila also show that Rag GTPases regulate cell growth, autophagy and animal viability during starvation. These studies establish a function of Rag GTPases in TORC1 activation in response to amino acid signals (Kim, 2008).
The multiprotein mTORC1 protein kinase complex is the central component of a pathway that promotes growth in response to insulin, energy levels, and amino acids and is deregulated in common cancers. This study finds that the Rag proteins [a family of four related small guanosine triphosphatases (GTPases)] interact with mTORC1 in an amino acid-sensitive manner and are necessary for the activation of the mTORC1 pathway by amino acids. A Rag mutant that is constitutively bound to guanosine triphosphate interacted strongly with mTORC1, and its expression within cells made the mTORC1 pathway resistant to amino acid deprivation. Conversely, expression of a guanosine diphosphate-bound Rag mutant prevented stimulation of mTORC1 by amino acids. The Rag proteins do not directly stimulate the kinase activity of mTORC1, but, like amino acids, promote the intracellular localization of mTOR to a compartment that also contains its activator Rheb (Sancak, 2008).
TOR complex 1 (TORC1) is a potent anabolic regulator of cellular growth and metabolism. When cells have sufficient amino acids, TORC1 is active due to its lysosomal localization mediated via the Rag GTPases. Upon amino acid removal, the Rag GTPases release TORC1, causing it to become cytoplasmic and inactive. This study shows that, upon amino acid removal, the Rag GTPases also recruit TSC2 to the lysosome, where it can act on Rheb. Only when both the Rag GTPases and Rheb are inactive is TORC1 fully released from the lysosome. Upon amino acid withdrawal, cells lacking TSC2 fail to completely release TORC1 from the lysosome, fail to completely inactivate TORC1, and fail to adjust physiologically to amino acid starvation. These data suggest that regulation of TSC2 subcellular localization may be a general mechanism to control its activity and place TSC2 in the amino-acid-sensing pathway to TORC1 (Demetriades, 2014).
TORC1, in Drosophila consisting of Target of rapamycin, Raptor and Lst8) regulates growth and metabolism, in part, by influencing transcriptional programs. This study has identified REPTOR and REPTOR-BP, both leucine zipper DNA-binding proteins, as transcription factors downstream of TORC1 that are required for approximately 90% of the transcriptional induction that occurs upon TORC1 inhibition in Drosophila. Thus, REPTOR and REPTOR-BP are major effectors of the transcriptional stress response induced upon TORC1 inhibition, analogous to the role of FOXO downstream of Akt. When TORC1 is active, it phosphorylates REPTOR on Ser527 and Ser530, leading to REPTOR cytoplasmic retention. Upon TORC1 inhibition, REPTOR becomes dephosphorylated in a PP2A-dependent manner, shuttles into the nucleus, joins its partner REPTOR-BP to bind target genes, and activates their transcription. In vivo functional analysis using knockout flies reveals that REPTOR and REPTOR-BP play critical roles in maintaining energy homeostasis and promoting animal survival upon nutrient restriction (Tiebe, 2015).
Target of rapamycin complex 1 (TORC1) integrates information on energy and nutrient status in eukaryotic cells. Under high-nutrient and -energy conditions, TORC1 drives translation, ribosome biogenesis, mitochondrial activity, lipid synthesis, nucleotide synthesis, and glycolysis. TORC1, thereby, couples activity of cellular anabolic and catabolic pathways to nutrient and energy supply. TORC1 is frequently mis-regulated in diseases such as cancer, diabetes, obesity, and neurodegeneration (Tiebe, 2015).
TORC1 regulates growth and metabolism by phosphorylating target proteins, such as S6K and 4E-BP, involved in translational regulation. Phosphorylation of targets changes very rapidly upon altered TORC1 activity, allowing cells to adapt quickly to changing environmental conditions. In addition, TORC1 also has long-lasting impact on cellular behavior through the control of transcriptional programs. This occurs by directly or indirectly modulating the activity of transcription factors such as SREBP, HIF1a, PGC-1a, TIF1a, PPARa, Atf4 (CREB2), TFEB, and TFE3 (Tiebe, 2015).
The TORC1 signaling pathway is highly conserved through evolution, thereby enabling the use of model organisms such as Drosophila for discovery of novel pathway components. Recent studies in Drosophila analyzed the impact of TORC1 signaling on cellular transcription. In Drosophila S2 cells, inhibition of TORC1 with rapamycin leads to numerous transcriptional changes. Genes involved in anabolic processes such as ribosome biogenesis are strongly repressed upon TORC1 inhibition. Previous work showed that this occurs via downregulation of myc activity (Teleman, 2008). A second class of genes is activated upon TORC1 inhibition. Although the function of these genes is less understood, they probably represent the genes needed for cells to adapt to conditions yielding reduced TORC1 activity, such as low nutrient availability. This study aimed to find the transcription factor responsible for mediating this upregulation upon TORC1 inhibition. The discovery is reported of these factors, which, surprisingly, are required for mediating most of the transcriptional induction that takes place upon TORC1 inhibition and play important roles in maintaining energy homeostasis in vivo (Tiebe, 2015).
This study has identify two uncharacterized genes, CG13624 and CG18619, which are termed REPTOR and REPTOR-BP, respectively, as transcription factors mediating circa 90% of the transcriptional repression downstream of TORC1 in Drosophila, which indicates that they are major effectors of TORC1. REPTOR is inhibited by TORC1-mediated phosphorylation and cytoplasmic retention by 14-3-3 proteins. Upon nutrient deprivation and low TORC1 activity, REPTOR becomes active, accumulating in the nucleus and binding target genes, a process that requires its partner, REPTOR-BP (Tiebe, 2015).
REPTOR is repressed when TORC1 activity is high, as is the case during larval stages when animals are feeding and growing. Hence, genetic removal of REPTOR during larval stages of well-fed animals has little phenotypic consequences, including no growth defects. In contrast, REPTOR is somewhat activated in (1) pupae and adults, which eat significantly less than larvae; (2) in larvae growing in low-nutrient conditions; and (3) in S2 cells growing in standard culture conditions. Hence, under these conditions, REPTOR loss of function leads to transcriptional and physiological phenotypes. The strongest REPTOR phenotypes become apparent when animals are starved, as these are the conditions where TORC1 is most inactive and, hence, REPTOR is most active (Tiebe, 2015).
The REPTOR regulatory system is analogous to another nutrient sensitive pathway-that of Akt and FOXO. When nutrients are low, Akt becomes inactive due to reduced systemic insulin signaling. This leads to FOXO dephosphorylation, release from 14-3-3 proteins, and nuclear accumulation, thereby activating target genes that mount a stress response. FOXO and REPTOR can be thought of as the respective counterparts for insulin and TORC1 signaling, which sense nutrients at the systemic and cell-autonomous levels, respectively. FOXO and REPTOR also have common target genes and bind near each other in shared enhancers. In sum, FOXO and REPTOR appear to have overlapping functions; indeed, genetic removal of both REPTOR and FOXO is synthetic lethal, indicating that they compensate for loss of each other (Tiebe, 2015).
As an effector of TORC1 function, REPTOR mediates some of the physiological effects of reduced TORC1 activity. Body-wide inhibition of TORC1 signaling leads to increased TAG levels and animals of reduced size. These phenotypes are, in part, due to activation of REPTOR, since removal of REPTOR strongly rescues them. Thus, TORC1 promotes growth during development not only by activating S6K but also by keeping REPTOR/REPTOR-BP repressed. Inhibition of TORC1 also extends lifespan. This could potentially be mediated, in part, via activation of REPTOR/REPTOR-BP, since both REPTOR and REPTOR-BP KO animals have significantly reduced lifespans (Tiebe, 2015).
Triglyceride levels were quantified in TOR2L1/2L19 hypomorphic larvae and found to be significantly elevated compared to those in controls, in line with a number of reports from adult flies. These results do not fit with one report that dTOR7/P mutant larvae are lean. The reason for this discrepancy or whether it has to do with the particular nature of the dTOR[7] and/or dTOR[P] alleles is unknown. Further work will be required to examine this carefully (Tiebe, 2015).
Both REPTOR and REPTOR-BP proteins have DBDs. Hence, the DNA-binding specificity of the REPTOR/REPTOR-BP dimer likely reflects the combined action of the two proteins. Since REPTOR-BP can also homodimerize, the REPTOR-BP homodimer might bind DNA with a pattern distinct from that of the REPTOR/REPTOR-BP dimer (Tiebe, 2015).
Many of the genes repressed by rapamycin in larvae (~80%) were no longer repressed by rapamycin in REPTOR KO larvae, raising the possibility that these genes are also REPTOR targets. It is not thought, however, that this is the case for several reasons. (1) In S2 cells, almost no genes require REPTOR to be repressed by rapamycin. If REPTOR were also involved in transcriptional repression, this would likely be seen also in S2 cells. (2) The REPTOR-induced genes are in common between S2 cells and larvae, whereas the REPTOR-repressed ones are not, suggesting their regulation might result from indirect effects. (3) Transactivation assays with REPTOR and REPTOR-BP only show strong transcriptional activation of the reporters but no repression. That said, many transcription complexes have both activating and repressive activities, so further investigation might find that REPTOR and REPTOR-BP also have repressive functions (Tiebe, 2015).
Surprisingly, REPTOR and REPTOR-BP have attracted little attention to date.
Microarray studies found that expression of CG13624 and CG18619 are regulated by nutrient conditions in the fly; however, no information regarding their function was available. Using BLAST to compare the human proteome with REPTOR and REPTOR-BP identifies Crebrf and Crebl2, respectively, which could potentially be human orthologs. It will be interesting to study them in light of the Drosophila data (Tiebe, 2015).
In summary, this study identifies REPTOR and REPTOR-BP as dedicated transcription factors that control the transcriptional repression downstream of TORC1 in Drosophila. Since these transcription factors mediate part of the functional output of TORC1, it will be interesting to assess their contribution toward the role that TORC1 plays in cancer, diabetes, and aging (Tiebe, 2015).
Glycolysis and fatty acid (FA) synthesis directs the production of energy-carrying molecules and building blocks necessary to support cell growth, although the absolute requirement of these metabolic pathways must be deeply investigated. This study used Drosophila genetics and focused on the TOR (Target of Rapamycin) signaling network that controls cell growth and homeostasis. In mammals, mTOR (mechanistic-TOR) is present in two distinct complexes, mTORC1 and mTORC2; the former directly responds to amino acids and energy levels, whereas the latter sustains insulin-like-peptide (Ilp) response. The TORC1 and Ilp signaling branches can be independently modulated in most Drosophila tissues. This study shows that TORC1 and Ilp-dependent overgrowth can operate independently in fat cells and that ubiquitous over-activation of TORC1 or Ilp signaling affects basal metabolism, supporting the use of Drosophila as a powerful model to study the link between growth and metabolism. Cell-autonomous restriction of glycolysis or FA synthesis in fat cells was shown to retrain overgrowth dependent on Ilp signaling but not TORC1 signaling. Additionally, the mutation of FASN (Fatty acid synthase) results in a drop in TORC1 but not Ilp signaling, whereas, at the cell-autonomous level, this mutation affects none of these signals in fat cells. These findings thus reveal differential metabolic sensitivity of TORC1- and Ilp-dependent growth and suggest that cell-autonomous metabolic defects might elicit local compensatory pathways. Conversely, enzyme knockdown in the whole organism results in animal death. Importantly, this study weakens the use of single inhibitors to fight mTOR-related diseases and strengthens the use of drug combination and selective tissue-targeting (Devilliers, 2021).
Adaptation to nutrient scarcity involves an orchestrated response of metabolic and signaling pathways to maintain homeostasis. This study found that in the fat body of fasting Drosophila, lysosomal export of cystine coordinates remobilization of internal nutrient stores with reactivation of the growth regulator target of rapamycin complex 1 (TORC1). Mechanistically, cystine was reduced to cysteine and metabolized to acetyl-coenzyme A (acetyl-CoA) by promoting CoA metabolism. In turn, acetyl-CoA retained carbons from alternative amino acids in the form of tricarboxylic acid cycle intermediates and restricted the availability of building blocks required for growth. This process limited TORC1 reactivation to maintain autophagy and allowed animals to cope with starvation periods. It is proposed that cysteine metabolism mediates a communication between lysosomes and mitochondria, highlighting how changes in diet divert the fate of an amino acid into a growth suppressive program (Parkhitko, 2022).
Maintaining cellular homeostasis upon nutrient shortage is an important challenge for all animals. Decreased activity of TORC1 is necessary to limit translation, reduce growth rates, and promote autophagy. Conversely, minimal TORC1 activity is required to promote lysosomal biogenesis, thus maintaining autophagic degradation necessary for survival. Using Drosophila as an in vivo model, this study found that TORC1 reactivation upon fasting integrates the biosynthesis of amino acids from anaplerotic inputs into the control of growth. The regulation of aspartate abundance appears to be critical during this process, possibly because it serves as a cataplerotic precursor for various macromolecules, including other amino acids and nucleotides, which in turn impinge on TORC1 activity. Cysteine recycling through the lysosome may fuel acetyl-CoA synthesis and prevent reactivation of TORC1 above a threshold that would compromise autophagy and survival during fasting. Reactivation of TORC1 during fasting was not passively controlled by the extent of amino acid remobilized from the lysosome. Instead, cysteine metabolism supported an increased incorporation of the carbons from these remobilized amino acids into the TCA cycle. It is therefore proposed that the remobilized amino acids may be transiently stored in the form of TCA cycle intermediates compartmentalized in the mitochondria, thereby restricting their accessibility. The regulation of TORC1 activity over a fasting period appears to be a combination of activating and suppressing cues that conciliate autophagy with anabolism. This process is self-regulated by autophagy, because autophagic protein degradation controls cystine availability through the lysosomal cystinosin transporter. Thus, in contrast to fed conditions, in which amino acid transporters at the plasma membrane maintain high cytosolic concentration of leucine and arginine that can directly be sensed by members of the TORC1 machinery, TORC1 reactivation in prolonged fasting is regulated indirectly by lysosome-mitochondrial cross-talk. Because cystinosin has also been shown to physically interact with several components of lysosomal TORC1 in mammalian cells, additional layers of regulation are conceivable during this process (Parkhitko, 2022).
Multiple functions of cysteine impinge on cellular metabolism, including transfer RNA thiolation, the generation of hydrogen sulfide, the regulation of hypoxia-inducible factor (HIF), and its antioxidant function through glutathione synthesis. Supplementation with cysteine or modified molecules such as N-acetyl-cysteine (NAC) can be used to efficiently buffer oxidative stress and perhaps alleviate symptoms of diseases that promote oxidative stress or glutathione deficiency, including cystinosis. Cysteine or NAC treatment extends the life span in flies, worms, and mice, and mice fed NAC show a sudden drop in body weight similar to that caused by dietary restriction. The results indicate that cysteine may not only act through its antioxidant function but also by restricting the availability of particular amino acids and limiting mTOR activity, processes known to extend life span. Moreover, this study shows that CoA is a main fate of cysteine that affects oxidative metabolism in the mitochondria, which is the main source of reactive oxygen species (ROS). Thus, the antioxidant function of cysteine also might be coupled to its effects on the mitochondria to buffer ROS production (Parkhitko, 2022).
In summary, this study demonstrate that cysteine metabolism acts in a feedback loop involving de novo CoA synthesis, the TCA cycle, and amino acid metabolism to limit TORC1 reactivation upon prolonged fasting. This pathway may be particularly important for developing organisms that must maintain autophagy and balance growth and survival during periods of food shortage (Parkhitko, 2022).
Animals can sense internal nutrients, such as amino acids/proteins, and are able to modify their developmental programs in accordance with their nutrient status. In the fruit fly, Drosophila melanogaster, amino acid/protein is sensed by the fat body, an insect adipose tissue, through a nutrient sensor, target of rapamycin (TOR) complex 1 (TORC1). TORC1 promotes the secretion of various peptide hormones from the fat body in an amino acid/protein-dependent manner. Fat-body-derived peptide hormones stimulate the release of insulin-like peptides, which are essential growth-promoting anabolic hormones, from neuroendocrine cells called insulin-producing cells (IPCs). Although the importance of TORC1 and the fat body-IPC axis has been elucidated, the mechanism by which TORC1 regulates the expression of insulinotropic signal peptides remains unclear. This study shows that an evolutionarily conserved molecular chaperone, heat shock protein 90 (Hsp90), promotes the expression of insulinotropic signal peptides. Fat-body-selective Hsp90 knockdown caused the transcriptional downregulation of insulinotropic signal peptides. IPC activity and systemic growth were also impaired in fat-body-selective Hsp90 knockdown animals. Furthermore, Hsp90 expression depended on protein/amino acid availability and TORC1 signaling. These results strongly suggest that Hsp90 serves as a nutrient-responsive gene that upregulates the fat body-IPC axis and systemic growth. It is proposed that Hsp90 is induced in a nutrient-dependent manner to support anabolic metabolism during the juvenile growth period (Ohhara, 2021).
Animals select food based on hungers that reflect dynamic macronutrient needs, but the hormonal mechanisms underlying nutrient-specific appetite regulation remain poorly defined. This study identified tachykinin (Tk) as a protein-responsive gut hormone in Drosophila and female mice, regulated by conserved environmental and nutrient-sensing mechanisms. Protein intake activates Tk-expressing enteroendocrine cells (EECs), driving the release of gut Tk through mechanisms involving target of rapamycin (TOR) and transient receptor potential A1 (TrpA1). In flies, this study delineated a pathway by which gut Tk controls selective appetite and sleep after protein ingestion, mediated by glucagon-like adipokinetic hormone (AKH) signalling to neurons and adipose tissue. This mechanism suppresses protein appetite, promotes sugar hunger and modulates wakefulness to align behaviour with nutritional needs. Inhibiting protein-responsive gut Tk prolongs lifespan through AKH, revealing a role for nutrient-dependent gut hormone signalling in longevity. These results provide a framework for understanding EEC-derived nutrient-specific satiety signals and the role of gut hormones in regulating food choice, sleep and lifespan (Ahrentlov, 2025).
The dysregulation of the metabolic regulator TOR complex I (TORC1) contributes to a wide array of human pathologies. Tuberous sclerosis complex (TSC) is a potent inhibitor of TORC1. This study demonstrates that the Rag GTPase acts in both the amino-acid-sensing and growth factor signaling pathways to control TORC1 activity through the regulation of TSC dynamics in HeLa cells and Drosophila. TSC lysosomal-cytosolic exchange increases in response to both amino acid and growth factor restriction. Moreover, the rate of exchange mirrors TSC function, with depletions of the Rag GTPase blocking TSC lysosomal mobility and rescuing TORC1 activity. Finally, this study shows that the GATOR2 complex controls the phosphorylation of TSC2, which is essential for TSC exchange. These data support the model that the amino acid and growth factor signaling pathways converge on the Rag GTPase to inhibit TORC1 activity through the regulation of TSC dynamics (Yang, 2020).
Cells must sense and adapt to a constantly changing external environment. Accordingly, in both single-celled and multicellular organisms, the ability to switch between anabolism, which supports growth, and catabolism, which is activated in response to stress, is critical to the maintenance of metabolic homeostasis. The highly conserved target of rapamycin (TOR) complex 1 (TORC1) is central to the regulation of metabolism in eukaryotes. TORC1 serves as a hub to integrate multiple upstream signaling inputs and regulates the execution of downstream metabolic pathways that control growth, proliferation, and cell death. TORC1 consists of five subunits including mTOR, which functions as a serine-threonine kinase, as well as Deptor, Raptor, mLST8, and PRAS40. In the presence of positive upstream inputs, TORC1 promotes anabolic metabolism by phosphorylating downstream effectors such as 4E-BP1 and S6K to increase mRNA translation and protein synthesis. Concurrently, TORC1 inhibits catabolic pathways such as autophagy, by phosphorylating essential autophagic proteins including ULK1 and Atg13. Thus, through the regulation of mTORC1 activity, cells can rapidly react to a diverse array of positive and negative environmental cues (Yang, 2020).
An important aspect of TORC1 regulation is its recruitment to and activation of lysosomes. These two processes require different Ras-related small GTPases, Rag and Rheb, respectively. In the presence of amino acids, TORC1 is translocated from the cytosol to the lysosomal membrane by the Rags. Specifically, RagA or RagB dimerizes with RagC or RagD to form a heterodimeric complex that recruits TORC1 to the lysosome by directly interacting with the Raptor subunit in an amino-acid-dependent manner. Once on the lysosome, Rheb binds to the subunit mTOR resulting in a conformational change that exposes active site residues allowing TORC1 to bind and phosphorylate a wide array of substrates (Yang, 2020).
As small GTPases, the functions of Rags and Rheb are regulated by their guanine nucleotide-binding cycle, which is tightly controlled by upstream signals. The Rags switch their TORC1-recruiting function by changing their guanine-nucleotide-binding status. When cells lack access to appropriate levels of amino acids, the GTPase-activating protein toward Rags 1 (GATOR1) complex blocks the function of TORC1 by acting as a GTPase-activating protein (GAP) toward RagA/B. Thus, under amino acid starvation the guanine-nucleotide-binding status of RagA/B changes to GDP inhibiting the ability of the Rag heterodimer to recruit TORC1 onto lysosomes. Another GATOR subcomplex, GATOR2, functions to oppose GATOR1 activity and is composed of five highly conserved proteins (Mio, Wdr24 (absent from Drosophila), Wdr59, Seh1, and Sec13), several of which have been shown to localize to lysosomes. In the presence of sufficient amino acids, the GATOR2 complex activates TORC1 activity by opposing the activity of the GATOR1 complex. The mechanistic details of how the GATOR2 complex opposes the activity of the GATOR1 complex to activate TORC1 remain unclear (Yang, 2020).
The small GTPase Rheb is inhibited by a three-subunit complex, called the tuberous sclerosis complex (TSC), comprised the proteins TSC2, TSC1, and TBC1D7. In the absence of insulin, TSC2 possesses a GAP activity toward Rheb. TSC serves as a potent inhibitor of the TORC1 signaling pathway. Mutations or deletions in TSC subunits result in the growth of benign tumors, epilepsy, and developmental delay. The upstream signals controlling TSC have been extensively studied. Essentially, growth factors, including insulin, activate class I phosphatidylinositol-3-kinase (PI3K), which in turn stimulates the protein kinase AKT. AKT inhibits TSC by directly phosphorylating multiple serine residues in TSC2. Thus, growth factors and insulin positively regulate the TORC1 signaling pathway by preventing TSC from inhibiting the TORC1 activator Rheb (Yang, 2020).
While the model that TSC controls TORC1 activity through the inhibition of Rheb is well established, the detailed mechanism of how TSC is regulated remains poorly understood. It is widely accepted that TSC responds to insulin and other growth factors through a PI3K-AKT circuit. However, the role of TSC in response to changes in amino acid levels is unclear. One possibility is that TORC1 is regulated by two independent pathways; the PI3K-AKT pathway that regulates the activity of the TORC1 inhibitor TSC in response to growth factors and the Rag GTPase pathway that responds to amino acid levels. However, multiple laboratories have reported an essential role for TSC in the response to amino acid starvation. Additionally, it is unclear exactly how intracellular TSC dynamics impact the ability of TSC to inhibit TORC1. Several studies have shown that TSC shuttles between lysosomes and the cytoplasm in response to amino acid availability, whereas others have reported that the lysosomal localization of TSC is independent of amino acid status with the TSC complex being released from lysosomes in response to high levels of insulin. Notably, in some cell types, such as HeLa cells, TSC is constitutively present on lysosomes even in the presence of physiological concentrations of insulin. Thus, whether the recruitment of TSC from the cytosol to the lysosomes is a common feature of TSC regulation in all cell types remains unclear. Finally, the detailed mechanism of how the phosphorylation of TSC2 by AKT affects TSC's function is not well understood. Several studies proposed that TSC2 undergoes proteasomal degradation after phosphorylation by AKT. However, more recent work suggests that TSC is not regulated at the level of protein stability but that the entire TSC complex is released from lysosomes upon phosphorylation of the TSC2 subunit by AKT. Thus, a full understanding of TSC regulation requires further exploration (Yang, 2020).
This study demonstrates that the regulation of TSC lysosomal dynamics by the Rag GTPase is required for the full response to both amino acid and growth factor restriction. Moreover, using fluorescence recovery after photobleaching (FRAP) and a photoconvertible fluorescently tagged TSC2, this study demonstrates that in response to negative stimuli, the Rag GTPase drives rapid cycling of TSC on and off lysosomes in both HeLa cells and Drosophila. Importantly, this study finds that GATOR2 and the Rag GTPase impact the lysosomal cycling of TSC2 by regulating its inhibitory phosphorylation by AKT. These data support the model that the Rag GTPase works in concert with the PI3/AKT/TSC pathway to regulate TORC1 activity in response to both amino acid and growth factor restriction (Yang, 2020).
TSC is a critical inhibitor of TORC1 signaling. Currently, there are two working models for the role of TSC in the inhibition of TORC1 activity. The first model posits that TSC lies exclusively downstream of the PI3K-AKT growth factor signaling pathway, while the second model proposes that TSC is a critical downstream effector of both the growth factor signaling and amino-acid-sensing pathways. The current findings on the function of the GATOR2 complex are consistent with the second model, which implicate different nucleotide states of the Rag GTPase in the recruitment of TORC1 versus TSC to lysosomes in response to amino acid starvation. Moreover, this study tried to find that the Rag GTPase, which has previously been thought to exclusively function in amino acid sensing, regulates the recruitment of TSC to lysosomes in response to growth factor restriction. Thus, the data support a model in which both the amino-acid-sensing pathway and growth factor signaling pathway converge on the Rag GTPase to recruit TSC to lysosomes in response to inhibitory signals. Notably, this study found in both HeLa cells and Drosophila, the Rag GTPase promotes the rapid exchange of TSC between the lysosome and cytosol in response to negative inputs. Finally, demonstrating further integration of the amino-acid-sensing and growth factor signaling pathways, this study shows that the GATOR2 complex acts upstream of the Rag GTPase to promote both, the activating phosphorylation of AKT and the AKT-dependent inhibitory phosphorylation of TSC2 (Yang, 2020).
An important outstanding question in the field of TORC1 regulation concerns the role of the Rag GTPase and TSC. The data indicate that in both Drosophila and HeLa cells, the GATOR-Rag GTPase axis inhibits TORC1 activity through the regulation of the dynamic behavior of TSC. In cells without a functional GATOR2 complex, the Rag GTPase, locked in its RAGAGDP: RAGCGTP-bound form due to the activation of GATOR1 recruits TSC to lysosomes precluding the recruitment and activation of TORC1. This inhibited state is relieved by depleting RAGA or RAGC, resulting in the recruitment and activation of TORC1 on lysosomes. Mutations in GATOR2 components mimic amino acid starvation. Thus, the current results are consistent with reports from the Teleman laboratory indicating that amino acid starvation promotes the Rag GTPase-dependent recruitment of TSC to lysosomes (Demetriades, 2014). Moreover, the current data confirm that under conditions of metabolic homeostasis TORC1 can be recruited to lysosomes and activated by Rheb in the absence of the Rag GTPase (Yang, 2020).
Intriguingly, in budding yeast, which does not have TSC, the homolog of the Rag GTPase, GTRl1/GTRL2, has a similar dual role in the regulation of TORC1 activity with the overexpression of GTR1GTP promoting TORC1 activity while GTR1GDP is associated with low TORC1 activity. Thus, the Rag GTPase may have a conserved role in the inhibition of TORC1 activity that goes beyond the regulation of TSC dynamics (Yang, 2020).
Surprisingly, this study also demonstrated a central role of the Rag GTPase in the recruitment of TSC to lysosomes in response to growth factor restriction. Specifically, the low TORC1 activity and reduced TSC mobility associated with growth factor depletion were rescued by depleting the Rag GTPase component RagA. Taken together, the data argue that the Rag GTPase and TSC are critical components of both the amino-acid-sensing and growth factor signaling pathways (Yang, 2020).
The idea that TSC acts downstream of the GATOR-Rag GTPase pathway is consistent with previous observations in Drosophila showing that depleting components of TSC rescue the low TORC1 activity observed in GATOR2 mutant ovaries. Indeed, in Drosophila, TSC is epistatic to the GATOR2 complex with respect to TORC1 activity, in that, depleting TSC in GATOR2 mutant cells resulted in high TORC1 levels similar to those observed in TSC single mutants. As indicated above, these data are also consistent with several previous reports that TSC acts in the amino-acid-sensing pathway (Yang, 2020).
Recent reports indicate that the recruitment of TSC to lysosomes is a common response to cellular stress in many mammalian cell types. However, these studies have not examined if TSC remains static on lysosomes or actively exchanges with the cytoplasmic pool during periods of TORC1 inhibition. Using FRAP and a photoconvertible TSC2 to follow the intracellular dynamics of TSC components, this study determined that the rate of exchange of TSC between the lysosome and the cytosol increases in response to nutrient deprivation and growth factor restriction. This rapid increase in cycling on and off lysosomes requires the Rag GTPase in both HeLa cells and the Drosophila ovary. Notably, increased cycling of TSC correlates with its increased interaction with Rheb and a concomitant decrease in TORC1 activity. Depleting components of the Rag GTPase in cells grown in amino acid or growth factor restricted media dramatically reduced the lysosomal-cytosol TSC rate of exchange, decreased its binding with Rheb, and rescued TORC1 activity. Thus, the data support the model that the Rag GTPase increases the exchange rate of TSC between the lysosome and the cytoplasm in response to both amino acid starvation and growth factor restriction (Yang, 2020).
It has long been established that AKT controls TSC activity through the inhibitory phosphorylation of TSC2. More recent studies indicate that AKT controls TSC activity by regulating its association with Rheb on the surface of lysosomes. This study finds that in nutrient-replete conditions, the inhibitory phosphorylation of TSC2 by AKT slows the rate of lysosomal-cytosolic exchange of TSC and allows for TORC1 activation. When TSC2 is rendered resistant to AKT-dependent phosphorylation, TSC rapidly cycled on and off lysosomes independent of growth factor and amino acid status. Importantly, cells expressing AKT-resistant TSC have low TORC1 activity. This study determined that in WDR24-KO cells, in which the RAG GTPase is in the RAGAGDP: RAGCGTP state, the AKT-dependent phosphorylation of TSC2 is strongly diminished, resulting in the increased mobility of the TSC complex on lysosomes and decreased TORC1 activity. Depleting components of the Rag GTPase in WDR24-KO cells, rescued these phenotypes resulting in increased AKT-dependent phosphorylation of TSC, decreased TSC lysosomal motility, and increased TORC1 activity. Further examination revealed that levels of activated AKT are kept low in WDR24-KO cells by a Rag GTPase-dependent mechanism. These data are consistent with previous observations that RAGA-KO cells have increased levels of activated AKT while WDR24-KO cells fail to increase AKT activation after Sestrin2 overexpression. Taken together, the data indicate that the GATOR2 complex promotes the activation of AKT, which facilitates the AKT-dependent inhibitory phosphorylation of TSC, upstream of the Rag GTPase. Moreover, they demonstrate that the rate of TSC cycling on and off lysosomes reflects TSC activity (Yang, 2020).
Based on these data, the following model is proposed. Under conditions of amino acid and growth factor sufficiency, GATOR2 inhibits GATOR1 resulting in the Rag GTPase adopting the RAGAGTP: RAGCGDP configuration, which favors the recruitment and activation of TORC1 on lysosomes and results in the limited exchange of TSC between the lysosome and the cytoplasm. In contrast, under conditions of amino acid or growth factor depletion, or in GATOR2 mutant cells, GATOR1 is active, resulting in the Rag GTPase adopting the RAGAGDP: RAGCGTP configuration, which promotes the rapid exchange of TSC between the lysosome and the cytoplasm and decreases TORC1 activity. Under these restricted conditions (AA−, serum−, or WDR24-KO), knockdowns of RAGA or RAGC prevent the rapid cycling of TSC allowing for the recovery of TORC1 activity due to the inherent ability of TORC1 to bind Rheb directly (Demetriades, 2014). Currently, whether the rapid cycling of TSC on and off lysosomes in response to upstream signals directly promotes TSC activity, or serves a regulatory role, has not been definitively established. In summary, these data support a model in which both the amino-acid-sensing and growth factor signaling pathways utilize the Rag GTPase to inhibit TORC1 activity through the regulation of TSC lysosomal dynamics (Yang, 2020).
A recent study reported that RAGA rapidly cycles between the lysosome and the cytosol in response to nutrients. Notably, RAGAGTP cycles on and of lysosomes while RAGAGDP remains more tightly associated with the Ragulator at the lysosomal surface. Intriguingly, this study found that RAGAGDP promotes the rapid cycling of the TSC complex on and off the lysosome. Thus, in the future, it will be important to determine precisely how the dynamic behavior of RAGA and TSC are coordinated to control TORC1 activity (Yang, 2020).
Amino acids regulate TOR complex 1 (TORC1) via two counteracting mechanisms, one activating and one inactivating. The presence of amino acids causes TORC1 recruitment to lysosomes where TORC1 is activated by binding Rheb. How the absence of amino acids inactivates TORC1 is less well understood. Amino acid starvation recruits the TSC1/TSC2 complex to the vicinity of TORC1 to inhibit Rheb; however, the upstream mechanisms regulating TSC2 are not known. This study identified the the eIF4A-containing eIF4F translation initiation complex (composed of three subunits: eIF4E, eIF4A and eIF4G) as an upstream regulator of TSC2 in response to amino acid withdrawal in Drosophila. TORC1 and translation preinitiation complexes bind each other. Cells lacking eIF4F components retain elevated TORC1 activity upon amino acid removal. This effect is specific for eIF4F and not a general consequence of blocked translation. This study identifies specific components of the translation machinery as important mediators of TORC1 inactivation upon amino acid removal (Tsokanos, 2016).
To maintain homeostasis, biological systems frequently use a combination of two distinct mechanisms that converge and counteract each other. For instance, the level of phosphorylation of a target protein depends not only on the rate of phosphorylation by the upstream kinase, but also on the rate of dephosphorylation by the phosphatase. Both the activating kinase and the inactivating phosphatase can be regulated separately. Likewise, the activity of TORC1 in response to amino acid levels appears to reflect a balance between activating and inactivating mechanisms that converge on Rheb. When amino acids are re-added to cells, TORC1 is activated via Rag or Arf1 GTPase-dependent recruitment to the lysosome where TORC1 binds Rheb (Kim, 2008; Sancak, 2008). In contrast, when amino acids are removed from cells, TORC1 activity drops in part by blocking this activation mechanism and in part via a distinct inactivation mechanism whereby TSC2 is recruited to the vicinity of TORC1 to act on Rheb (Demetriades, 2014). The existence of this distinct and counteracting mechanism is highlighted by the fact that in the absence of TSC2, both Drosophila and mammalian cells do not appropriately inactivate TORC1 in response to amino acid removal (Demetriades, 2014). The upstream mechanisms regulating TSC2 in response to amino acid withdrawal, however, are not known. This study has identified the translational machinery, and in particular components of the eIF4F complex, as one upstream regulatory mechanism working via TSC2 to inactivate TORC1 upon amino acid withdrawal (Tsokanos, 2016).
The subcellular localization of TORC1 plays an important role in its regulation. A significant body of evidence shows that TORC1 needs to translocate to the lysosome or Golgi to become reactivated following amino acid starvation and re-addition. Whether active TORC1 then remains on the lysosome, or whether it can move elsewhere in the cell to phosphorylate target proteins, is less clear. Several findings in the literature, as well as the data presented in this study, indicate that active TORC1 can leave the lysosome, yet remain active: (1) Upon amino acid re-addition in starved cells, the Rag GTPases are necessary for mTORC1 lysosomal localization and reactivation. In contrast, Rag depletion in cells growing under basal conditions, replete of serum and amino acids, does not cause a strong drop in mTORC1 activity, although it causes a similar delocalization of mTORC1 away from lysosomes. Hence, under these conditions, mTORC1 is non-lysosomal, but still active to a large extent. (2) Similarly, particular stresses such as arsenite treatment can cause TORC1 to localize away from the lysosome, yet remain active. (3) The Rag GTPases tether TORC1 to the LAMTOR complex present on the lysosome. Amino acid restimulation, which activates TORC1, actually decreases binding between Rag GTPases and LAMTOR, suggesting that active Rag-bound TORC1 complexes can leave the lysosome and reside elsewhere in the cell. Additional mechanisms also contribute to the delocalization of the Rag GTPases away from lysosomes (4) Active TORC1 phosphorylates target proteins such as 4E-BP and S6K, which are physically associated with translation preinitiation complexes. Indeed, this study reports physical interactions between the TORC1 complex and translation preinitiation complexes, in agreement with what has also been observed by others. Therefore, either translation preinitiation complexes need to translocate to lysosomes to meet TORC1, or TORC1 needs to come off the lysosome to meet translation preinitiation complexes in the cytoplasm. (5) Using proximity ligation assay, an interaction was observed between Raptor and eIF4A, which does not colocalize with either lysosomes or endoplasmic reticulum, suggesting that it takes place in the cytoplasm. (6) In agreement with these PLA data, antibody staining of cells in the presence of amino acids with anti-TOR antibody reveals an accumulation of TOR on lysosomes, as well as a more diffuse, non-lysosomal TORC1 localization throughout the cytoplasm. (7) A recent report employing a FRET-based probe detects mTORC1 activity at lysosomes as well as in the cytoplasm and nucleus. Taken together, these data suggest that although TORC1 is activated on the lysosome, it then in part translocates to other sites in the cell including the cytoplasm to phosphorylate target proteins (Tsokanos, 2016).
Upon amino acid withdrawal, both cytoplasmic and lysosomal fractions of active TORC1 need to be inactivated. The data presented in this study suggest that upon amino acid removal, inactivation of TORC1 happens in part via an eIF4A-dependent mechanism acting on TSC2 to inactivate Rheb in the cytosol. In agreement with this, TORC1 inactivation upon amino acid removal can be rescued by supplying cells with dominantly active, but not wild-type Rheb. It has been previously reported that a pool of TSC2 is also recruited to lysosomes upon amino acid removal (Demetriades, 2014). This study shows in Drosophila cells, upon amino acid removal, some TSC2 accumulates in lysosomes, whereas some remains in the cytosol. Therefore, TSC2 is likely recruited to all subcellular sites where active TORC1 is located to inactivate it. Indeed, Rheb and TSC2 have been observed at several subcellular compartments. Since Rheb localizes to many endomembranes in the cell, Rheb that is not bound to TORC1 could potentially remain active, to provide a pool for subsequent TORC1 reactivation (Tsokanos, 2016).
Upon inactivation, the data indicate that TORC1 remains bound to preinitiation complexes, in agreement with previous reports. This finding is reminiscent of the fact that Raptor is also recruited to stress granules, which are essentially stalled preinitiation complexes, in response to another stress-oxidative stress. Whether the Rag GTPases also remain bound to preinitiation complexes upon amino acid removal is unclear because some experiments showed a decrease in binding between Rag GTPases and initiation factors, and some did not (Tsokanos, 2016).
How could eIF4A affect TORC1 activity? The data indicate that the effects of eIF4A knockdown cannot be explained as a consequence of generally impaired translation, since other means of blocking translation do not have the same effects on TORC1 activity upon amino acid starvation. Instead, knockdown of any of the three members of the eIF4F complex gives this elevated TORC1 phenotype, indicating that it is specific for the eIF4F complex. The data are consistent with two interpretations: One option is that the eIF4F complex is specifically required to translate a protein that promotes TSC2 function. An alternate option is that the eIF4F complex acts directly on TSC2, regulating its activity. The latter is supported by the fact that eIF4A and TSC2 proteins are seen interacting with each other. Interestingly, eIF4A has been reported to have additional functions that are not translation-related (Tsokanos, 2016).
Some differences were noted between Drosophila cells and mammalian cells. The first is that overexpression of wild-type Rheb is sufficient to activate TORC1 upon amino acid removal in mammalian cells, whereas this is not the case in Drosophila cells. This could be due to a difference in the biology of the two cell types, or simply to a technical difference having to do with levels of Rheb overexpression. A second difference is that cycloheximide treatment is sufficient to maintain elevated TORC1 levels in HeLa or HEK293 cells upon amino acid removal, whereas this is not the case in Drosophila cells. This could be due to differences in rates of amino acid efflux and levels of autophagy in mammalian compared to S2 and Kc167 cells, causing intracellular amino acid levels to remain elevated in mammalian cells when both amino acid import from the medium and amino acid expenditure via translation are simultaneously blocked (Tsokanos, 2016).
A number of studies have looked at the involvement of Rheb in the cellular response to amino acids, with some disagreement on whether amino acids affect Rheb GTP-loading or Rheb-mTOR binding. The current data fit with previous reports that Rheb GTP-loading is affected by amino acids and with the conclusion that amino acids affect TORC1 activity via both a Rheb-dependent and a Rheb-independent mechanism (Tsokanos, 2016).
The data indicate a close physical relationship between TORC1 and the translational machinery. This is in part mediated by a direct interaction between the major scaffolding subunit of the initiation complex, eIF4G, and RagC and in part likely mediated by additional interactions between TORC1 and preinitiation supercomplexes as previously reported. Interestingly, TORC2 is also physically associated with the ribosome and requires ribosomes, but not translation, for its activation. Hence, both TORC1 and TORC2 have close physical connections to the translational machinery (Tsokanos, 2016).
Some side observations in this study are interesting and could constitute a starting point for further studies. For instance, eIF4A-knockdown cells inactivate TORC1 more robustly than control cells upon serum removal. Also, eIF2b knockdown causes S6K phosphorylation to decrease significantly in S2 cells. It is not known why this occurs. The latter might suggest that there are additional points of cross-talk between TORC1 and the translation machinery (Tsokanos, 2016).
How cells sense the presence or the absence of amino acids has been an open question in the field. The data presented in this study indicate that the translational machinery itself might sense the absence of amino acids. Indeed, the relevant parameter for a cell is likely not the absolute levels of intracellular amino acids, but rather whether the available amino acid levels are sufficient to support the amount of translation that a cell requires. Hence, the translation machinery itself might be best poised to make this assessment. Binding is observed between eIF4A and NAT1 that is strong in the presence of amino acids, and is reduced upon amino acid withdrawal, independently of TORC1 signaling. These epistasis experiments are consistent with NAT1 acting as the upstream mediator of the amino acid signal, binding and inhibiting eIF4A in the presence of amino acids, but not in the absence of amino acids. Hence, NAT1 might play a role in this sensing process (Tsokanos, 2016).
In sum, these data identify the eIF4F complex as an important upstream regulator of TORC1, which acts via TSC2 to inactivate TORC1 upon withdrawal of amino acids (Tsokanos, 2016).
Stem cells reside in niches that provide signals to maintain self-renewal, and differentiation is viewed as a passive process that depends on losing access to these signals. This study demonstrates that differentiation of somatic cyst stem cells (CySCs) in the Drosophila testis is actively promoted by PI3K/Tor signaling, as CySCs lacking PI3K/Tor activity cannot properly differentiate. An insulin peptide produced by somatic cells immediately outside of the stem cell niche was found to act locally to promote somatic differentiation through Insulin receptor (InR) activation. These results indicate that there is a local 'differentiation' niche which upregulates PI3K/Tor signaling in the early daughters of CySCs. Finally, it was demonstrated that CySCs secrete the Dilp-binding protein ImpL2, the Drosophila homolog of IGFBP7, into the stem cell niche, which blocks InR activation in CySCs. Thus, this study shows that somatic cell differentiation is controlled by PI3K/Tor signaling downstream of InR and that local production of positive and negative InR signals regulate the differentiation niche. These results support a model in which leaving the stem cell niche and initiating differentiation is actively induced by signaling (Amoyel, 2016).
This study shows that PI3K/Tor activity is required for the differentiation of somatic stem cells in the Drosophila testis. Additionally, a 'differentiation' niche was identified immediately adjacent to the stem cell niche that,
through the local production of Dilps, leads to the upregulation of PI3K/Tor activity in early CySC daughters and to their commitment to differentiation. The secretion of ImpL2 by CySCs antagonizes the initiation of differentiation in CySCs by blocking available Dilps in the stem cell niche. As a result, CySCs receive little free Dilp ligands. However, as their daughters move away from the hub, they encounter increasing levels of Dilps and decreasing levels of ImpL2, which leads to the upregulation of PI3K/Tor signaling and proper somatic cell differentiation. The fact that ImpL2
is upregulated by the main self-renewal signal (i.e., JAK/STAT) in CySCs leads to a model accounting for the spatial separation of the stem cell niche and the differentiation niche (Amoyel, 2016).
The results are consistent with a model in which autocrine or paracrine production of Dilp6by early cyst cells serves as a differentiation niche in the testis, defining where in the tissue upregulation PI3K/Tor signaling - a prerequisite for differentiation - occurs. This differentiation niche is critical for somatic development because stem cell markers like Zfh1 are maintained in the absence of signals like PI3K/Tor. Notably, JAK/STAT activity is not expanded outside of the niche upon somatic loss of PI3K/Tor signaling, suggesting that differentiation signals play a critical role in downregulating stem cell factors. Intriguingly, recent studies in the Drosophila ovary have identified a differentiation niche in this tissue: autocrine Wnt ligands produced by somatic support escort cells regulate escort cell function, proliferation and viability. Taken together, these studies reveal that at least in Drosophila gonads, there is a defined region immediate adjacent to the stem cell niche where autocrine production of secreted factors induces the differentiation of somatic cells, which in turn promote development of the germ line (Amoyel, 2016).
Several studies have examined the role of insulin signaling in gonadal stem cells. In both testes and ovaries, systemic Dilps have been shown to affect stem cell behavior. In both tissues, nutrition through regulation of systemic insulin controls the proliferation rate of GSCs. The current data showing that Akt1, Dp110 or Tor mutant CySC clones proliferate poorly are consistent with these findings and indicate that basal levels of insulin signaling are required for the proliferation and/or survival of both stem cell pools in the testis. This work also demonstrates that production of a secreted Insulin binding protein ImpL2 by CySCs reduces available Dilps in the stem cell niche, and ImpL2 in the niche milieu should reduce insulin signaling in GSCs and CySCs. While these data seemingly contradict the results that insulin is required for GSC maintenance, a model is suggested in which low constitutive levels of insulin signaling are required for stem cell proliferation and that higher levels are required to induce stem cell differentiation. (Amoyel, 2016).
Prior reports have found that both male and female flies with reduced Insulin or Tor activity are sterile, and the results presented in this study suggest that this is due at least in part to a lack of somatic cell differentiation. The results indicate that Dilp6, the IGF homolog, plays a local role in CySC differentiation, but acts redundantly with other presumably systemic factors, suggesting that both constitutive and nutrient-responsive inputs control CySC differentiation. Indeed, this study shows that in addition to controlling the proliferation of stem cells, systemic insulin is required for their differentiation, as the poorly proliferative Akt1, Dp110 or Tor mutant CySC clones do not differentiate and eventually die by apoptosis. This combination of reduced proliferation and increased apoptosis may explain why other studies suggest that Tor is required for self-
renewal in GSCs; indeed prior reports indicate that while Tor mutant GSCs are lost, hyper-activation of Tor leads to faster loss of GSCs through differentiation and recent work indicates that lineage-wide Tor loss blocks the differentiation of GSCs. The use of hypomorphic alleles enabled a genetic separation of the proliferative effects and differentiation requirements of PI3K and Tor in CySCs. Finally, there is evidence that PI3K/Tor activity promotes differentiation of stem cells in gonads in mammals, suggesting that these findings may reflect a conserved role of Tor activity in promoting germ cell differentiation, both through autonomous and non-
autonomous mechanisms involving somatic support cells. Moreover, it seems likely that Tor activity may be a more general requirement for the differentiation of many stem cell types, as increased PI3K or Tor has been shown to induce differentiation in many instances. In particular, mouse long term hematopoietic stem cells are lost to differentiation when the PI3K inhibitor Pten
is mutated, while Drosophila intestinal stem cells differentiate when Tor is hyperactive due to Tsc1/2 complex inactivation. Moreover, inhibition of Tor activity by Rapamycin promotes cellular reprogramming to pluripotency, while cells with increased Tor activity cannot be reprogrammed, suggesting a conserved role for Tor signaling in promoting differentiated states (Amoyel, 2016).
The balance between self-renewal and differentiation ensures long-term maintenance of stem cell (SC) pools in regenerating epithelial tissues. This balance is challenged during periods of high regenerative pressure and is often compromised in aged animals. This study shows that target of rapamycin (TOR) signaling is a key regulator of SC loss during repeated regenerative episodes. In response to regenerative stimuli, SCs in the intestinal epithelium of the fly and in the tracheal epithelium of mice exhibit transient activation of TOR signaling. Although this activation is required for SCs to rapidly proliferate in response to damage, repeated rounds of damage lead to SC loss. Consistently, age-related SC loss in the mouse trachea and in muscle can be prevented by pharmacologic or genetic inhibition, respectively, of mammalian target of rapamycin complex 1 (mTORC1) signaling. These findings highlight an evolutionarily conserved role of TOR signaling in SC function and identify repeated rounds of mTORC1 activation as a driver of age-related SC decline (Haller, 2017).
The mechanistic (or mammalian) target of rapamycin complex 1 (mTORC1) controls cell growth, proliferation, and metabolism in response to diverse stimuli. Two major parallel pathways are implicated in mTORC1 regulation including a growth factor-responsive pathway mediated via TSC2/Rheb and an amino acid-responsive pathway mediated via the Rag GTPases. This study identified and characterize three highly conserved growth factor-responsive phosphorylation sites on RagC, a component of the Rag heterodimer, implicating cross talk between amino acid and growth factor-mediated regulation of mTORC1. RagC phosphorylation is associated with destabilization of mTORC1 and is essential for both growth factor and amino acid-induced mTORC1 activation. Functionally, RagC phosphorylation suppresses starvation-induced autophagy, and genetic studies in Drosophila reveal that RagC phosphorylation plays an essential role in regulation of cell growth. Finally, mTORC1 was identified as the upstream kinase of RagC on S21. These data highlight the importance of RagC phosphorylation in its function and identify a previously unappreciated auto-regulatory mechanism of mTORC1 activity (Yang, 2018).
mTOR is an evolutionarily conserved atypical serine/threonine kinase belonging to the phosphoinositide 3 kinase (PI3K)-related kinase family. mTOR is found in two structurally and functionally distinct complexes-mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2)-defined by their unique components, in particular raptor (mTORC1) and rictor (mTORC2). Through the coordinated phosphorylation of its downstream effectors, mTORC1 integrates extra- and intra-cellular signal inputs such as amino acids, growth factors (GF), stress, and energy status, to regulate major cellular processes including growth, proliferation, and survival. Underlining its crucial role in cellular and organismal homeostasis, mTORC1 dysregulation occurs in numerous human diseases including cancer, metabolic disorders, and neurodegeneration. Growth factors and amino acids both acutely enhance mTORC1 activity, and two different types of small GTPases-Ras-homolog enriched in brain (Rheb) and the Rag GTPases-cooperatively regulate mTORC1 activity via these two parallel activation mechanisms. Rheb is activated under conditions of high cellular ATP and upstream growth factor signals. Once activated, Rheb interacts with and activates mTORC1 and is required for mTORC1 activation by all signals, including amino acids. Rag GTPases are considered amino acid-specific regulators of the mTORC1 pathway. Mammals have four Rag proteins-RagA to RagD-which form obligate heterodimers comprising RagA or RagB together with RagC or RagD. Amino acids cause Rag GTPases to switch to an active conformation, in which RagA/B is GTP-loaded and Rag C/D is GDP-loaded. The active Rag heterodimer physically interacts with raptor, recruiting mTORC1 to the lysosome where its activator Rheb resides. Extensive work has revealed several mechanisms implicated in the regulation of Rag activity that enables them to function as nutrient sensors. A common feature among these is the control of Rag nucleotide status, particularly through the activation of guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). These include the Ragulator (GEF for Rag A/B; Bar-Peled, 2012), the GATOR1 complex (GAP for RagA/B; Bar-Peled, 2013), folliculin (FLCN, GAP for RagC/D; Petit, 2013; Tsun, 2013), and leucyl-tRNA synthetase (LeuRS, GAP for RagD; Han, 2012). Ubiquitination has also recently emerged as a post-translational modification (PTM) capable of inhibiting Rag GTPase signaling by recruiting GATOR1 to RagA. Importantly, these pathways regulating Rag activity are all amino acid-dependent, and much less is known about the control of growth factor-mediated Rag GTPase signaling (Yang, 2018).
In a recent global mass spectrometry-based phosphoproteomics study in adipocytes, insulin-dependent phosphorylation was observed of several highly conserved residues on RagC including S2, S21, and T394. These data highlight a possible role for the Rag GTPases in mTORC1 growth factor sensing. This study demonstrates that both growth factors and amino acids trigger RagC phosphorylation and that phosphorylated RagC potentiates mTORC1 activity and affects mTORC1-dependent cell growth and autophagy. Moreover, the phosphorylation of RagC at S21 (and likely T394) was shown to be catalyzed directly by mTORC1, revealing a novel auto-regulatory feedback loop within the mTORC1 signaling pathway (Yang, 2018).
This study identified a new auto-regulatory branch of mTORC1 signaling, involving phosphorylation of the Rag GTPase RagC. This is the first report that Rag GTPase phosphorylation can regulate mTORC1 activity. More importantly, the results confirm that Rag GTPases are not only involved in the amino acid-sensing mTORC1 pathway, but could also participate in growth factor sensing in the mTORC1 pathway. Although previous studies show that in Rag heterodimers, the GTP/GDP loading of Rag heterodimers plays a dominant role in the interaction between Rag heterodimers and mTORC1, the data indicate that RagC is also a positive regulator of mTORC1 through post-translational modification. Interestingly, phosphoproteomics data suggest that most phosphorylation is concentrated on RagC compared with other Rag GTPases, and S21 is not conserved between RagC and RagD, suggesting that RagC is not functionally redundant and potentially has distinct biological functions to RagD (Yang, 2018).
One of the RagC phosphorylation sites, S21, was established as a novel rapamycin-insensitive mTORC1 substrate in vitro and in cells, and the T394 is phosphorylated by mTOR in vitro. The S2 and T394 sites may also be mTORC1 substrates in vivo, because the kinetics of their phosphorylation resembles that of other bona fide mTORC1 substrates and they also have surrounding sequence features matching the preferred sequence motif of mTORC1. These findings indicate the presence of a positive feedback loop between mTORC1 and RagC, which may contribute to the fine-tuning of mTORC1 activity (Yang, 2018).
There is evidence that the stability of the raptor-mTOR complex is related to mTORC activity, and the current data implicate RagC phosphorylation in the destabilization of mTORC1. This is likely to be a direct effect of RagC phosphorylation, because RagC 3E still destabilized mTOR-raptor complex under serum starvation. This is consistent with the observation that RagC 3E causes hyper-phosphorylation of ULK1 and inhibits autophagy under serum starvation. The next major question is what is the underlying cause of this instability. One possibility is that RagC phosphorylation influences the interaction with other regulators, resulting in 'locking' or 'opening' of the mTOR-raptor complex. Interestingly, it was observed that RagC 3A binds more FLCN, which is a GAP for RagC/D, and RagC 3E can bind more raptor under both steady and amino acid starvation/re-fed condition. One possibility is that RagC phosphorylation regulates its nucleotide binding status by modulating the interaction with FLCN. However, no substantial difference was observed in FLCN binding between wild-type RagC and RagC 3E, or raptor binding between wild-type RagC and 3A. The temporal change in mTOR/raptor stability upon amino acid re-feeding is similar to that of FLCN/RagC stability, but not with raptor/RagC interaction. A possible explanation is that FLCN has two functions: serving as a GAP for RagC/D and a 'lock' for mTORC1. This model could help explain why FLCN releases from Rag GTPases in the presence of amino acids if it is a GAP for RagC/D, which is a positive regulator for mTORC1 activity: After activating RagC/D, FLCN needs to be disassociated from the lysosome to unlock mTORC1, and RagC phosphorylation may affect this process. Further studies will be needed to investigate these possibilities (Yang, 2018).
Other explanations cannot be ruled out for the impact of RagC phosphorylation on impaired mTORC1 activity. For example, it is well established that raptor recruits substrate proteins such as S6K and 4E-BP1 to mTORC1 so that they can be phosphorylated by mTOR. Therefore, RagC phosphorylation may affect the recruitment of mTORC1 substrates by raptor. Recently, two elegant studies showed that under amino acid or growth factor starvation, the Rag heterodimer binds and recruits TSC2 to lysosomes to inhibit Rheb, resulting in mTORC1 inactivation. Therefore, a final possibility is that RagC phosphorylation may mediate its effects by acting through TSC2. Future studies into the underlying mechanics of how RagC phosphorylation exerts its effects on mTORC1 signaling are therefore likely to shed light on this newly identified mechanism that sits at the intersection between amino acid sensing and growth factor signaling (Yang, 2018).
mTOR Complex (mTORC1) promotes cell growth and proliferation in response to nutrients and growth factors. Amino acids induce lysosomal translocation of mTORC via the Rag GTPases. Growth factors activate Ras homolog enriched in brain (Rheb), which in turn, activates mTORC at the lysosome. Amino acids and growth factors also induce the phospholipase D (PLD)-phosphatidic acid (PA) pathway, required for mTORC signaling through mechanisms that are not fully understood. Using human and murine cell lines, along with immunofluorescence, confocal microscopy, endocytosis, PLD activity, and cell viability assays, this study shows that exogenously supplied PA vesicles deliver mTORC to the lysosome in the absence of amino acids, Rag GTPases, growth factors, and Rheb. Of note, pharmacological or genetic inhibition of endogenous PLD prevented mTORC lysosomal translocation. This study observed that precancerous cells with constitutive Rheb activation through loss of TSC complex subunit (TSC2) exploit the PLD-PA pathway and thereby sustain mTORC activation at the lysosome in the absence of amino acids. These findings indicate that sequential inputs from amino acids and growth factors trigger PA production required for mTORC translocation and activation at the lysosome (Frias, 2019).
mTORC18 is a conserved serine/threonine catalytic complex that integrates signals from nutrients and growth factors to regulate cell growth, proliferation, survival, and metabolism. Activation of mTORC1 is a two-step process whereby amino acids induce Rag-dependent translocation of mTORC1 from the cytoplasm to the lysosome, followed by mTOR kinase activation by the lysosomal small GTPase Rheb upon growth factor stimulation (Frias, 2019).
Phospholipase D (PLD) and its product, the signaling lipid phosphatidic acid (PA) play a role in mTORC1 activation in response to amino acids and growth factors. Amino acids induce lysosomal translocation of PLD1. Once on the lysosome, PLD1 binds to Rheb, which activates PLD1 in response to growth factors. PLD1 is widely expressed in mammals and converts the most abundant membrane phospholipid phosphatidylcholine to choline and PA. Conserved basic amino acids in the FKBP12-rapamycin binding (FRB) domain of mTOR lead to proton dissociation to generate PA with two negative charges. This locks mTOR onto deprotonated PA, promoting mTORC1 assembly and stability (Frias, 2019).
This study reports that PA with an unsaturated fatty acid stimulates lysosomal translocation and activation of mTORC1 in the absence of amino acids, Rag GTPases, growth factors, or Rheb. This work provides a unifying model showing that PA is critical for translocation and full activation of mTORC1 at the lysosome in response to sequential signals provided by amino acids and growth factors (Frias, 2019).
The data support a model where PLD1, similar to mTORC1, acts like a coincidence detector and effector of both amino acids and growth factors. Amino acids induce PLD activity and production of PA. Exogenously supplied PA vesicles enter the cell through endocytosis and drive mTOR to the lysosome, suggesting that amino acids induce production of PA-containing endosomes that carry mTOR to the lysosome. In agreement, inhibition of endogenous PLD prevented mTOR translocation to the lysosome in response to amino acids. Amino acid-induced RagA/B-GTP RagC/D-GDP heterodimers provide a parallel pathway that locks mTOR on the lysosome. Amino acids also induce the translocation of PLD1 from cytoplasmic puncta to the lysosome. Once on the lysosome, PLD1 binds to Rheb. Growth factors activate Rheb, which then activates PLD1. Lysosomal PA production promotes further binding of PA to mTOR to allow complex stability and activation (Frias, 2019).
Previous studies showed that exogenously supplied PA induced mTORC1 activation in the presence but not in the absence of amino acids. This study was able to induce mTORC1 translocation to the lysosome and mTORC1 activity with exogenously supplied PA-18:1 vesicles in the absence of amino acids. The main difference between the two studies is that this study performed amino acid and serum deprivation (to prevent contamination of amino acids present in serum) for 1 h, followed by amino acid stimulation for 10 min. In contrast, the previous study performed amino acid starvation for 2 h after overnight serum starvation, followed by amino acid stimulation for 30 min (Frias, 2019).
Previous findings suggest that mTORC1 assembles before reaching the lysosome because binding of mTOR to the Rag GTPases requires the mTORC1 component raptor. PA promotes mTORC1 assembly and stability. PA-containing endosomes carrying mTORC1 to the lysosome is therefore an attractive model in which PA would allow mTORC1 formation and stability. This study showed that exogenously supplied PA can drive mTOR to the lysosome and induce mTORC1 activity in TSC2-null MEFs where RagC and D were genetically ablated. Therefore, it is proposed that delivery of mTORC1 to the lysosome does not require the Rags. However, this study found that residual retention of mTOR at the lysosome in TSC2-null MEFs was lost upon RagC and D knockdown. This suggests that the Rags operate in parallel to PA to lock mTORC1 on the lysosome, after mTORC1 delivery by PA. This study showed that genetic ablation of PLD1 induced lysosomal scattering. This favors the idea that PA-containing endosomes carry mTORC1 to the lysosome, fuse with the lysosome, and increase its size (Frias, 2019).
Exogenously supplied PA vesicles induce mTORC1 translocation and activation in the absence of Rheb, indicating that the key step in Rheb activation of mTORC1 is increased PLD1 activity and PA production. Consistent with this observation, the Rheb association with mTOR is independent of GTP loading, whereas the Rheb association with PLD1 depends on GTP loading. Additionally, this study found that PLD1/2 inhibitors, in combination, abolished mTORC1 activity in RagAGTP/GTP MEFs, indicating that PA production is required for complex assembly and stability. If mTORC1 is not intact, then constitutive Rag activation is lost. Thus, the effect of PA on mTORC1 is downstream of Rheb and parallel to Rag GTPases.
Genetic deletion of mTOR in mice is embryonic lethal. Unlike mTOR, mice with genetic deletion of PLD1, PLD2, or both are viable, suggesting that PLD and PA may not be required for steady state but rather acute activation of mTOR. PLD inhibitors preferentially killed TSC-null MEFs, whereas rapamycin did not. This suggests an advantage in terms of cancer therapeutics because targeting PLD may selectively target cancer cells, with minimal side effects. PLD inhibitors were developed from halopemide, a psychotropic drug extensively used in humans without toxicities. Thus, PLD inhibition might be a viable alternative to current therapies in cancers with mTORC1 hyperactivation that requires PLD-generated PA (Frias, 2019).
The TORC regulator GATOR1/SEACIT controls meiotic entry and early meiotic events in yeast. However, how metabolic pathways influence meiotic progression in metazoans remains poorly understood. This study examined the role of the TORC regulators GATOR1 and GATOR in the response to meiotic double-stranded breaks (DSB) during Drosophila oogenesis. In mutants of the GATOR component mio, meiotic DSBs trigger the constitutive downregulation of TORC activity and a permanent arrest in oocyte growth. Conversely, in GATOR mutants, high TORC activity results in the delayed repair of meiotic DSBs and the hyperactivation of p53. Unexpectedly, it was found that GATOR inhibits retrotransposon expression in the presence of meiotic DSBs in a pathway that functions in parallel to p53. Thus, these studies have revealed a link between oocyte metabolism, the repair of meiotic DSBs and retrotransposon expression (Wei, 2019).
Metabolism impacts meiotic progression during oogenesis. Target of Rapamycin Complex 1 (TORC1) is a multi-protein complex that functions as a master regulator of metabolism. In the presence of adequate nutrients and positive upstream growth signals, TORC1, which contains the serine/threonine kinase Target of Rapamycin, becomes active and functions to stimulate growth and inhibit catabolic metabolism through the phosphorylation of down-stream effector proteins. The Seh1 Associated Complex Inhibits TORC1 (SEACIT), originally identified in yeast, inhibits TORC1 activity in response to amino acid limitation. SEACIT, known as the GAP Activity Towards Rags complex 1 (GATOR1) in metazoans, is comprised of three highly conserved proteins Npr2/Nprl2, Npr3/Nprl3 and Iml1/Depdc5. In Drosophila and mammals, depleting any of the three GATOR1 components results in increased TORC1 activity and growth, as well as a reduced response to amino acid starvation. Thus, the role of the SEACIT/GATOR1 complex in the regulation of TORC1 activity is highly conserved in eukaryotes (Wei, 2019).
The multi-protein GATOR2 complex, known as Seh1 Associated Complex Activates TORC1 (SEACAT) in yeast, inhibits the activity of GATOR1 and thus functions to activate TORC1 (see Mio prevents the constitutive downregulation of TORC1 activity in response to meiotic DSBs). In metazoans, the GATOR2 complex functions in multiple amino acid sensing pathways. In tissue culture cells, depleting GATOR2 components results in the constitutive activation of GATOR1 and the permanent downregulation of TORC1 activity. However, genetic studies of the role of individual GATOR2 components in Drosophila, indicate that the requirement for the GATOR2 complex is more nuanced when examined in the context of a multicellular animal. For example, mutations in the GATOR2 component mio, result in a block to oocyte growth and differentiation, due to the constitutive downregulation of TORC1 activity in the female germline. However, mio is not required to maintain TORC1 activity in most somatic tissues of Drosophila (Wei et al., 2016). Why there is a tissue specific requirement for mio in the female germline of Drosophila is currently unknown (Wei, 2019).
In single celled eukaryotes, nutrient limitation often facilitates meiotic entry. In the yeast Saccharomyces cerevisiae, the down-regulation of TORC1 by SEACIT/GATOR1 in response to amino acid stress promotes both meiotic entry and early meiotic progression. Surprisingly, as is observed in yeast, during Drosophila oogenesis the GATOR1 complex promotes meiotic entry. These data raise the intriguing possibility that in Drosophila the GATOR1 complex and low TORC1 activity may be critical to the regulation of additional events of the early meiotic cycle (Wei, 2019).
This study reports that the GATOR complex is critical to the response to meiotic DSB during Drosophila oogenesis. Restraining TORC1 activity via a pathway that involves both GATOR1 and the Tuberous sclerosis complex (TSC) promotes the timely repair of meiotic DSBs and prevents the hyperactivation of p53 in the female germline. Notably, the delayed repair of meiotic DSBs in GATOR1 mutants is due, at least in part, to the hyperactivation of the TORC1 target S6K. Conversely, the data indicate that the GATOR2 component Mio opposes the activity of GATOR1 in the female germline, thus preventing the constitutive downregulation of TORC1 activity and allowing for the growth and development of the oocyte in later stages of oogenesis. Thus, this study has identified a regulatory loop required to modulate TORC1 activity in response to meiotic DSBs during Drosophila oogenesis. Finally, during the course of these studies, it was observed that the GATOR1 complex prevents the derepression of retrotransposon expression in the presence of meiotic DSBs (Wei, 2019).
Previous work has shown that in Drosophila, mutations in the GATOR2 component mio, result in the constitutive activation of the GATOR1 pathway in the female germline but not in somatic tissues. This study demonstrates that the tissue specific requirement for mio during oogenesis is due, at least in part, to the generation of meiotic DBSs during oogenesis. In Drosophila, only the female germline undergoes meiotic recombination and thus experiences the genotoxic stress associated with developmentally programmed DSBs. This study shows that in mio mutants, blocking the formation of meiotic DSBs prevents the constitutive downregulation of TORC1 activity thus allowing for the growth and development of the oocyte. These data are consistent with the model that meiotic DSBs trigger the activation of a TORC1 inhibitory pathway that must be opposed and/or attenuated by the GATOR2 component Mio (see A working model for the role of the GATOR complex in the response to meiotic DSBs) (Wei, 2019).
While there are several possible models that might explain these data, it is believed the most parsimonious explanation for these results is that the TORC1 inhibitory pathway activated by meiotic DSBs, involves both GATOR1 and TSC. This model is consistent with the ability of both GATOR1 and TSC depletions to rescue the mio mutant phenotype. Additionally, recent reports indicate that GATOR1 and TSC act in a common pathway to downregulate TORC1 activity in response to multiple upstream inhibitory inputs. Previously, it was determined that in Drosophila, amino acid starvation induces a dramatic GATOR1/TSC dependent decrease in TORC1 activity in somatic tissues, that far exceeds any reduction in TORC1 activity observed in GATOR2 null mutants. This observation strongly suggests that, in addition to the removal of the GATOR2 inhibition of GATOR1, there is an activation step that is required to fully potentiate the GATOR1/TSC pathway (Wei, 2019).
Thus, based on these data the following model is proposed. Meiotic DSBs activate, or are required to maintain, a GATOR1/TSC dependent pathway that downregulates TORC1 activity in the female germline. The GATOR2 component Mio is required to oppose or turnoff this pathway to prevent the constitutive downregulation of TORC1 activity in later stages of oogenesis. While it is believed that the data support the role of the GATOR1/TSC pathway, it is conceded that an alternative regulator of TORC1 activity may also be critical to the downregulation of TORC1 activity in response to meiotic DSBs (Wei, 2019).
Hyperactivation of TORC1 has been linked to defects in the DNA damage response in single celled and multicellular organisms. The observation that meiotic DSBs likely promote the GATOR1 dependent downregulation of TORC1 activity during Drosophila oogenesis, suggested that limiting TORC1 activity may be important to the regulation of meiotic DSB repair. In previous work, it was found that GATOR1 mutant ovaries had TORC1 activity levels approximately three times higher than those observed in wild-type ovaries. This study demonstrates that GATOR1 mutant ovaries exhibit multiple phenotypes consistent with the misregulation of meiotic DSB repair including, an increase in the steady state number of Mei-W68/Spo-11 induced DSBs, the retention of meiotic DSBs into later stages of oogenesis and the hyperactivation of p53. Importantly, RNAi depletions of Tsc1 partially phenocopied the GATOR1 ovarian defects. Thus, the misregulation of meiotic DSBs observed in GATOR1 mutant oocytes are due to high TORC1 activity and not to a TORC1 independent function of the GATOR1 complex (Wei, 2019).
Epistasis analysis between the GATOR1 component nprl3 and the Rad51 homolog spnA, strongly suggest that GATOR1 impacts the repair, rather than the generation, of meiotic DSBs. This study determined that double mutants of nprl2 and the Rad51 homolog spnA, which is required for the repair of meiotic DSBs, have approximately the same number of DSBs as spnA single mutants. These data are consistent with GATOR1 and spnA influencing the common process of DNA repair and are inconsistent with GATOR1 mutants producing supernumerary breaks (Wei, 2019).
These observations on the role of the GATOR1 complex during Drosophila oogenesis are particularly intriguing in light of similar meiotic defects observed in a npr3 mutants in Saccharomyces cerevisiae. In the sporulation proficient strain SK1, npr3 mutant cells enter meiosis and express the transcription factor and master regulator of gametogenesis IME1 with wild-type kinetics. Subsequently, npr3 mutants exhibit a mild delay in the generation of meiotic DNA breaks, but a substantial delay in the repair of meiotic DSBs. Thus, yeast and Drosophila SEACIT/GATOR1 mutants share a common meiotic phenotype, the delayed repair of meiotic DSBs. These results raise the intriguing possibility that low TORC1 activity may be a common feature of the early meiotic cycle in many organisms (Wei, 2019).
Notably, the data indicate that the delay in the repair of meiotic DSBs in GATOR1 mutants is due to the hyperactivation of the TORC1 downstream target S6K. S6K is a critical downstream effector of TORC1 that impacts multiple essential cellular processes including, but not limited to cell growth, energy balance and aging. Intriguingly, in mammals, S6K has been implicated in the regulation of the DNA damage response with hyperactivation of the TORC1-S6K pathway resulting in the accumulation of unrepaired DSBs and genome instability. Thus, similar to what is reported in mammals, the data are consistent with the model that the hyperactivity of the TORC1/S6K axis delays the repair of DSBs in Drosophila (Wei, 2019).
Finally, it was determined that GATOR1 mutants have a diminished response to DSBs outside the female germline in somatic tissues of Drosophila. Similar to what is observed in TSC mutant cells in humans that have increased levels of TORC1 activity, this study find that GATOR1 mutant embryos have a reduced ability to survive low levels of γ-irradiation. Moreover, in the somatic follicle cells of the ovary a delay was observed in the repair of DSBs after adult females are exposed to low levels of γ-irradiation. Thus, in Drosophila inappropriately high TORC1 activity delays the repair of DSBs in both the germline and somatic tissues (Wei, 2019).
The initiation of homologous recombination through the programmed generation of DNA double-stranded breaks (DSBs) is a universal feature of meiosis. DSBs represent a dangerous form of DNA damage that can result in dramatic and permanent changes to the germline genome. To minimize this destructive potential, the generation and repair of meiotic DSBs is tightly controlled in space and time . The activation of transposable elements represents an additional threat to genome integrity in germ line cells. Genotoxic stress, resulting from DNA damage, has been implicated in the deregulation of transposons in multiple organisms. Thus, germ line cells may be at an increased risk for transposon derepression due to the genotoxic stress associated with meiotic recombination. Consistent with this hypothesis, germ line cells have evolved extensive surveillance systems to detect and silence transposons beyond the pathways present in most somatic tissues (Wei, 2019).
Previous studies have shown that DNA damage promotes the deregulation of retrotransposon in multiple organisms, including Drosophila. In line with these studies, this study found that in GATOR1 mutants, the DSBs that initiate meiotic recombination trigger the deregulation of retrotransposon expression. Similarly, p53 mutant females derepress retrotransposon expression during oogenesis, but as observed in GATOR1 mutants, primarily in the presence of meiotic DSBs. Double mutants of nprl3, p53 exhibit a dramatic increase in retrotransposon expression relative to either p53 or nprl3 single mutants, implying that p53 and GATOR1 act through independent pathways to repress retrotransposon expression in the female germline. One possibility is that both GATOR1 and p53 independently impact genome stability. Thus, disabling both pathways may have an additive effect on both genome stability and retrotransposon expression. Consistent with the hypothesis that genome instability drives retrotransposon expression, this study found that mutants in spnA/Rad51, which fail to repair meiotic DSBs, also exhibit increased transcription of multiple retrotransposons. Intriguingly, the SpnA homolog Rad51, as well as other genes required for DNA repair, was recently identified in a high throughput screen for genes that suppress (Long Interspersed Element-1) LINE1 expression in mammalian tissue culture cells (Wei, 2019).
However, the current data also suggest that the GATOR1 complex may influence retrotransposon expression independent of the regulation of TORC1 activity. While both GATOR1 and TSC are required for the efficient repair of meiotic DSBs, in contrast to GATOR1 mutant ovaries, little to no increase was observed in retrotransposon expression in the Tsc1 depleted ovaries. We believe this reflects the incomplete depletion of Tsc1 by RNAi resulting in a reduced retention of meiotic DSBs relative to GATOR1 mutants. However, a second possibility is that the GATOR1 complex inhibits retrotransposon expression independent of TORC1 inhibition. As is observed with spnA the depletion of GATOR1 components, but not TSC components result in the activation of LINE1 expression in HeLa cells. Taken together, these data hint that the GATOR1 complex may impact retrotransposon expression in the germline via two independent pathways: First by promoting the repair of meiotic DSBs through the downregulation of TORC1 activity and second via a pathway that functions independent of TORC1 inhibition (Wei, 2019).
Genes encoding components of the GATOR1 complex are often deleted in cancers. As is observed in GATOR1 mutants, cancer cells frequently have increased TORC1 activity, increased genomic instability and increased retrotransposon expression. Thus, in the future it will be important to identify the molecular mechanism by which the GATOR1 complex influences both the response to genotoxic stress and the expression of retrotransposons under both normal and pathological conditions (Wei, 2019).
The mechanistic target of rapamycin-mLST8-raptor complex (mTORC1) functions as a central regulator of cell growth and metabolism in response to changes in nutrient signals such as amino acids. SAMTOR is an S-adenosylmethionine (SAM) sensor, which regulates the mTORC1 activity through its interaction with the GTPase-activating protein activity toward Rags-1 (GATOR1)-KPTN, ITFG2, C12orf66 and SZT2-containing regulator (KICSTOR) complex. This paper reports the crystal structures of Drosophila melanogaster SAMTOR in apo form and in complex with SAM. SAMTOR comprises an N-terminal helical domain and a C-terminal SAM-dependent methyltransferase (MTase) domain. The MTase domain contains the SAM-binding site and the potential GATOR1-KICSTOR-binding site. The helical domain functions as a molecular switch, which undergoes conformational change upon SAM binding and thereby modulates the interaction of SAMTOR with GATOR1-KICSTOR. The functional roles of the key residues and the helical domain are validated by functional assays. These structural and functional data together reveal the molecular mechanism of the SAM sensing of SAMTOR and its functional role in mTORC1 signaling (Tang, 2022).
The mechanistic target of rapamycin (mTOR) is a multifunctional kinase that plays important roles in embryonic development, aging, tumorigenesis, diabetes, and neurodegenerative diseases. In mammalian cells, mTOR forms two functionally distinct complexes, the mTOR-mLST8-Raptor complex (mTORC1) and the mTOR-mLST8-Rictor complex (mTORC2). In response to environmental conditions of energy, nutrients, and extracellular growth factors, mTORC1 modulates the anabolic pathway and promotes the initiation and elongation of protein translation through directly phosphorylating specific substrates such as S6 kinase 1 (S6K1) and 4E binding protein 1 (4EBP1). In addition, mTORC1 suppresses the catabolic pathway through inhibition of autophagy and lysosome biogenesis (Tang, 2022).
Two complementary parallel signaling pathways work together to render full activation of mTORC1 at the lysosomal membrane. On one hand, amino acids induce the conversion of small guanosine triphosphatases (GTPases) Ras-related GTP-binding protein A (RagA) to RagD to the active nucleotide-bound state, i.e., the guanosine triphosphate (GTP)-loaded state of RagA/B and the guanosine diphosphate-loaded state of RagC/D. After that, a lysosomal multisubunit machinery comprising the vacuolar-type adenosine triphosphatase (v-ATPase), the pentameric Ragulator complex, and the active Rag GTPases recruits mTORC1 to the lysosomal surface. On the other hand, the growth factor-stimulated kinase Akt phosphorylates and then inhibits the tuberous sclerosis complex, which acts as a GTPase-activating protein (GAP) for the small GTPase Rheb at the lysosomal membrane, where the active GTP-bound Rheb can fully activate mTORC1 (Tang, 2022).
Rag GTPases function as obligate heterodimers such that RagA/B interacts with RagC/D through their C-terminal roadblock domains, and their N-terminal GTPase domains dictate their interactions with the mTORC1 unique component Raptor. The GATOR1 complex, comprising three subunits [Nprl2, Nprl3, and DEP domain-containing protein 5 (DEPDC5)], functions upstream of Rag GTPases as a GAP for RagA/B to inactivate mTORC1 when amino acids are deficient. The KICSTOR scaffolding complex, consisting of four subunits [Kaptin, integrin-alpha FG-GAP repeat-containing protein 2 (ITFG2), C12orf66, and SZT2], tethers GATOR1 to the lysosomal surface and facilitates the interaction between GATOR1 and Rag GTPases. The GATOR2 complex, consisting of five subunits [WD repeat domain 59 (WDR59), WD repeat domain 24 (WDR24), meiosis regulator for oocyte development (MIOS), SEH1 like nucleoporin (SEH1L), and SEC13], functions upstream of GATOR1 as an inhibitor of GATOR1 and thus a positive regulator of mTORC1. The cytoplasmic leucine and arginine activate mTORC1 through regulating the dynamic interplay of GATOR1 and GATOR2. Upon leucine/arginine deprivation, the cytoplasmic leucine sensors Sestrin1/2 and SAR1B or the arginine sensor CASTOR1 (cytosolic arginine sensor for mTORC1 subunit 1) interact with GATOR2 and block the GATOR1-GATOR2 interaction, releasing the GAP activity of GATOR1 for RagA/B, and the binding of leucine to Sestrin1/2 and SAR1B or arginine to CASTOR1 impairs the sensor's interaction with GATOR2, leading to inactivation of GATOR1 and thus activation of mTORC1 (Tang, 2022).
Similar to leucine and arginine, methionine regulates mTORC1 in a Rag GTPase-dependent manner. However, the direct cytoplasmic methionine sensor in mTORC1 signaling has not been found so far. Recently, an S-adenosylmethionine (SAM)-binding protein SAMTOR (or C7orf60) was identified as a negative regulator of mTORC1, which functions upstream of Rag GTPases, GATOR1, and KICSTOR: SAMTOR can interact with GATOR1 to prompt the function of GATOR1 and/or KICSTOR in the absence of SAM, and suppresses Rag GTPases and mTORC1; with the supply of SAM, the binding of SAM to SAMTOR disrupts the interaction of SAMTOR with GATOR1-KICSTOR, leading to the inhibition of the GATOR1 GAP activity and, thus, the activation of mTORC1. SAM is synthesized from methionine and adenosine triphosphate (ATP), and the cellular SAM level is directly correlated with methionine. The silencing of methionine adenosyltransferase MAT2A, which catalyzes the synthesis of SAM from methionine and ATP, decreases the expression of SAMTOR and the activation of mTORC1. Loss of SAMTOR prevents the inhibition of mTORC1 caused by methionine starvation. Thus, SAMTOR serves as a cytoplasmic SAM sensor in the SAM/methionine-mediated mTORC1 signaling (Tang, 2022).
To illuminate the molecular mechanism of the functional role of SAMTOR in the SAM/methionine-mediated mTORC1 signaling, this study determined the crystal structures of Drosophila melanogaster SAMTOR (dSAMTOR) in apo and SAM- and S-adenosyl-l-homocysteine (SAH)-bound forms. Structural analysis shows that SAMTOR comprises an N-terminal helical domain and a C-terminal class I SAM-dependent methyltransferase (MTase) domain. The ligand (SAM/SAH) binds to the MTase domain and makes extensive hydrogen-bonding and hydrophobic interactions with the surrounding residues. The functional roles of the key residues involved in ligand binding are validated by mutagenesis and biochemical assays. In addition, it was found that the N-terminal helical domain exhibits a high flexibility and acts as a molecular switch in response to SAM/SAH binding. In the absence of SAM/SAH, the helical domain is positioned away from the ligand-binding site, allowing SAMTOR to interact with GATOR1-KICSTOR. The binding of SAM/SAH appears to induce conformational change of the helical domain to cover the ligand-binding site, blocking the interaction of SAMTOR with GATOR1-KICSTOR. The structural and functional data together provide insight into the molecular mechanism of SAM/SAH sensing by SAMTOR in the SAM/methionine-mediated mTORC1 signaling (Tang, 2022).
The sulfur-containing amino acid methionine presents a key metabolite in many aspects of mammalian physiology, including translation, epigenetics, cell proliferation, and various signaling pathways. In metazoans, the mTORC1 signaling pathway senses the cellular level of the methionine metabolite SAM rather than methionine through the SAM sensor SAMTOR to modulate the anabolic and catabolic metabolisms. This work determined the crystal structures of the MTase domain of dSAMTOR in SAM- and SAH-bound forms and the full-length V66W/E67P mutant dSAMTOR in apo form. Structural analyses reveal that dSAMTOR comprises an N-terminal helical domain and a C-terminal MTase domain. The helical domain consists of three α helices (αA to αC) and exhibits a high flexibility. The MTase domain adopts a class I MTase fold supplemented with some auxiliary structure elements. In the SAM- and SAH-bound dSAMTOR MTase structures, the SAM and SAH bind to the MTase domain in a similar binding manner and with comparable binding affinities. The MTase domain of dSAMTOR and the full-length dSAMTOR exhibit comparable binding affinities with the ligands, suggesting that the helical domain is not directly involved in ligand binding. Comparison of the SAM-bound MTase domain and the apo V66W/E67P mutant structures shows that ligand binding does not induce notable conformational changes in the overall structure of the MTase domain except a few minor conformational changes at the ligand-binding site. The ligand makes extensive hydrophilic and hydrophobic interactions with several conserved residues, and mutations of these residues impair ligand binding in vitro. Selective mutations with deficient ligand binding also abolish the SAM-sensing ability of hSAMTOR and the mutants could bind to GATOR1-KICSTOR both in the absence and in the presence of SAM in vivo. These results reveal the molecular mechanism of SAM sensing and binding by SAMTOR (Tang, 2022).
Recently, several direct amino acid sensors upstream of mTORC1 including the arginine sensor CASTOR1 and the leucine sensor Sestrins2 have been identified and the crystal structures of these sensors have been solved. The structural and functional data together have revealed the molecular mechanisms about how CASTOR1 and Sestrins2 sense and bind the ligands. However, as ligand binding does not induce substantial conformational changes of the proteins, the molecular mechanisms about how these sensors function as molecular switches upon ligand binding to interact with the downstream partners and then further regulate mTORC1 signaling remain unclear. On the basis of the structural and functional data of SAMTOR together with the structural comparison with the AlphaFold predicted SAMTOR structure and the structures of several MTases, a working model is proposed for how SAMTOR functions as a molecular switch upon SAM binding to regulate mTORC1 signaling as follows. The C-terminal MTase domain of SAMTOR contains both the ligand-binding site and the GATOR1-KICSTOR-binding site. The MTase domain of SAMTOR alone has the ligand binding ability as well as the GATOR1-KICSTOR binding ability in both the presence and absence of the ligand. In other words, it has the ability to sense the SAM level but lacks the switch function to regulate mTORC1 signaling upon SAM binding. On the other hand, while the N-terminal helical domain of SAMTOR is not required for the interactions with the ligand and GATOR1-KICSTOR, it has a high flexibility and functions as a molecular switch to turn on and off the binding ability of the MTase domain with GATOR1-KICSTOR and subsequently mTORC1 signaling. In the absence of SAM, the helical domain is positioned distantly from the ligand-binding site with a flexible conformation, and hence the potential binding site for GATOR1-KICSTOR is exposed and the MTase domain can interact with GATOR1-KICSTOR, leading to the inhibition of mTORC1 signaling. When SAM binds to SAMTOR, the helical domain is positioned on the top of the ligand-binding site and the conformation is stabilized by SAM binding, and hence the potential binding site for GATOR1-KICSTOR is blocked and thus the MTase domain cannot interact with GATOR1-KICSTOR, leading to the activation of mTORC1 signaling (Tang, 2022).
Biochemical and structural data show that dSAMTOR can bind SAH and SAM with comparable binding affinities, and SAH binds to the MTase domain with almost identical interactions with the surrounding residues as SAM. The previous cell biology data also show that like SAM, the binding of SAH to hSAMTOR can disrupt the interaction of hSAMTOR with GATOR1-KICSTOR and inhibit the GATOR1 GAP activity in mTORC1 signaling. As the concentrations of SAM and SAH in cells are estimated to be at similar levels, these results suggest that SAH might be able to regulate the function of SAMTOR in mTORC1 signaling in the same manner as SAM (Tang, 2022).
Previous studies show that the arginine sensor CASTOR1 consists of four tandem ACT domains and structurally resembles the C-terminal allosteric domains of aspartate kinases, suggesting that CASTOR1 may have evolved from the ancient aspartate kinase but lost the N-terminal kinase domain during evolution. The leucine sensor Sentrin2 is a homolog of bacterial reductase Ahpd but lacks a conserved cysteine required for the catalytic activity. So far, whether Senstrin2 has a reductase activity remains elusive. SAMTOR contains a conserved class I MTase domain, which is usually known to bind and exert the catalytic activity on DNA or RNA during the regulation of gene expression, repair of mutations, and protection against bacterial restriction enzymes. Sequence alignment shows that the SAMTOR proteins have a moderate sequence similarity (~11% identity) to Saccharomyces cerevisiae Bmt2, an MTase responsible for the m1A modification of 25S ribosomal RNA (rRNA). Structural comparison of dSAMTOR with the small RNA MTase Hen1 (PDB code 3HTX) and the long noncoding RNA MTase MePCE (methyl phosphate capping enzyme) (PDB code 6DCB) of class I MTases shows that both Hen1 and MePCE have a positively charged surface cleft around the active site of the MTase domain to bind the RNA substrate. In contrast, the MTase domain of dSAMTOR contains no such kind of positively charged surface cleft; instead, the electrostatic surface around the ligand-binding site is largely negatively charged or hydrophobic, which is apparently unfavorable for the binding of an RNA or DNA substrate, suggesting that SAMTOR is unlikely an RNA or DNA MTase. Nevertheless, whether SAMTOR has an MTase activity on proteins remains elusive and warrants further study (Tang, 2022).
In all eukaryotic organisms, the control of growth, metabolism, reproduction, and lifespan is realized by interactions of genetic and environmental signals. An important player in the regulatory network is the target of rapamycin (TOR) signaling pathway, which is triggered by nutritional cues. Given the pivotal role of TOR in regulating multiple processes in organisms, this study inhibited TOR by inducible expression of specific RNAi in Drosophila intestinal stem and progenitor cells or progenitor cells alone. TOR inhibition in stem and progenitor cells shortened the lifespan on both regular diet and under malnutrition. Moreover, flies became more short-lived under starvation or oxidative stress conditions if TOR was inhibited. TOR-RNAi expression resulted in a decrease in body glycogen and TAG levels. All these physiological and metabolic changes might be partially explained by significant changes in mRNA levels for genes encoding the Drosophila insulin-like peptides (dilp2, dilp3 and dilp5) with subsequent effects on insulin signaling to modulate gene expression in peripheral tissues (e.g. tobi and pepck transcripts). In the gut, a strong increase in transcript levels of cytokines upd2, upd3 and downstream target socs36e of the JAK/STAT signaling pathway in the gut indicate an important role for this signaling pathway when TOR is inhibited (Strilbytska, 2020).
Mitochondrial dysfunctions belong amongst the most common metabolic diseases but the signalling networks that lead to the manifestation of a disease phenotype are often not well understood. This study identified the subunits of respiratory complex I, III and IV as mediators of major signalling changes during Drosophila wing disc development. Their downregulation in larval wing disc leads to robust stimulation of TOR activity, which in turn orchestrates a complex downstream signalling network. Specifically, after downregulation of the complex I subunit ND-49 (mammalian NDUFS2), TOR activates JNK to induce cell death and ROS production essential for the stimulation of compensatory apoptosis-induced proliferation within the tissue. Additionally, TOR upregulates Notch and JAK/STAT signalling and it directs glycolytic switch of the target tissue. These results highlight the central role of TOR signalling in mediating the complex response to mitochondrial respiratory dysfunction and they provide a rationale why the disease symptoms associated with respiratory dysfunctions are often alleviated by mTOR inhibitors (Perez-Gomez, 2020).
Mitochondria play an essential function in cellular energetic and NADH metabolism. Five protein complexes (complex I-V) of the electron transport chain (ETC) have distinct functions in the oxidation of NADH and/or FADH2, maintenance of the inner mitochondrial membrane potential and production of ATP via oxidative phosphorylation. Moreover, they serve as signalling hubs for specific cellular events including ROS-mediated signalling, apoptosis and Ca2+ signalling. Mutations in mitochondrial enzymes are the most frequent metabolic mutations present in human (Perez-Gomez, 2020).
Complex I of the ETC is the node point in the mitochondrial NADH metabolism as it mediates electron transfer from NADH to the other respiratory complexes. Therefore, complex I inhibitors have been exploited as therapeutic targets in cancer treatment, although the mechanism of action is often unclear. On the other hand, complex I inhibition can lead to increased proliferation, depending on the cell type and complex I inhibitor used. Despite the fact that mitochondrial electron transport chain disorders are one of the most common human genetic diseases, the mechanisms behind the dichotomy in the functional outputs after complex I inhibition are not well understood (Perez-Gomez, 2020).
The mTOR pathway (TOR in Drosophila) is the key integrator of cellular metabolic inputs that connects cell growth with environmental signals, including nutrient and growth factors availability. It promotes cell growth by stimulation of cellular translation, anabolic metabolism and by inhibiting autophagy. At the same time, mTOR activation can lead to apoptosis in certain contexts. The upregulation of mTOR is observed during epithelial wound healing, during aging as well as in many types of cancers. Although strong inhibition of mitochondrial respiration can cause a metabolic catastrophe and cell death connected with mTOR inhibition through the activation of AMPK, increasing evidence suggest that many types of respiratory dysfunctions are connected with increase in mTOR activity and mTOR inhibition leads to alleviation of the phenotype (Perez-Gomez, 2020).
This study found that downregulation of mitochondrial respiratory complex I, III or IV stimulates TOR activity that directs major downstream signalling events, including Notch activation, metabolic changes and apoptosis-driven proliferation. As TOR overactivation balances between stimulation of apoptosis and proliferation, presented in this study suggests a possible mechanism for the observations when complex I inhibition promotes either cell death or proliferation in different contexts. The signalling network that was identified also suggests a possible explanation why the disease symptoms associated with respiratory dysfunctions are often alleviated by mTOR inhibitors (Perez-Gomez, 2020).
Despite the fact that mutations in mitochondrial enzymes are the most frequent metabolic mutations present in human, manifested in a whole range of clinical disorders, the actual signalling networks that are triggered by malfunctioning mitochondria to develop clinical symptoms are still not well understood. The results argue that TOR pathway is the key signalling effector triggered after downregulation of complex I, III or IV in the Drosophila wing disc. TOR activity in turn activates JNK, Notch and JAK/STAT signalling, boosts glycolysis and promotes compensatory apoptosis-induced proliferation to produce profound effects on tissue size and patterning (Perez-Gomez, 2020).
By placing TOR at the top of the signalling network triggered by complex I dysfunction, this study provides a rationale for numerous observations where TOR inhibition alleviated the disease symptoms associated with mitochondrial dysfunction. For example, the maternally inherited Leigh syndrome (MILS), caused by mutation in complex I subunit Ndufs4, is associated with enhanced mTOR activity in neurons and the disease symptoms can be alleviated using chemical mTOR inhibitors. Upregulation of mTORC1 has also been described as a key component of the mitochondrial integrated stress response during mitochondrial myopathy. Alongside this line, the low survival rate of flies with mutation in the ND2 subunit of complex I can also be rescued by chemical TOR inhibition in Drosophila. Moreover, an aggressive phenotype of breast cancer that is associated with complex I mutations can be reversed via restoration of complex I function that is associated with decreased mTOR activity. Therefore, the data showing the role of TOR at the very apex of the signalling hierarchy after complex I dysfunction makes it interesting to test if similar regulatory mechanism underpins other types of mitochondrial dysfunction (Perez-Gomez, 2020).
Respiratory inhibitors are used to surpress various types of cancers although respiratory dysfunction can also promote cancer progression. Based on the current data it can be hypothesized that the types of complex I dysfunctions that stimulate cancer progression would correlate with overstimulation of mTOR activity that initiates downstream signalling events promoting apoptosis but also apoptosis-induced proliferation. This may seem contradictory as complex I inhibition usually leads to decrease in ATP:ADP ratio that in turn activates AMPK, a known suppressor of mTOR activity. However, the evidence that complex I inhibition would actually lead to downregulation of mTOR activity is surprisingly scarce and concerns mainly complex I inhibitors like biguanides (metformin, fenformin) or fenofibrate. On the contrary, there is ample of observations where mTOR is increased during mitochondrial dysfunction, supporting the results. In fact, mTOR activity can be stimulated even in the presence of active AMPK, as in the case of the Leigh syndrome caused by mutation in complex I subunit Ndufs4. By balancing between stimulation of apoptosis and proliferation, the mTOR driven signalling network identified in this study may suggest a possible mechanism for the contradictory observations where complex I inhibition was reported to promote cell death but also support proliferation depending on context (Perez-Gomez, 2020).
One remaining question is how TOR is upregulated by mitochondria. One possibility may be an activation of TOR via mitochondrial Akt signalling and TOR complexes located in mitochondria-associated endoplasmic reticulum. However, since TOR operates at the top of the signalling hierarchy after complex I downregulation, it can also be speculated that its activity could be sensitive to the primary metabolic misbalance caused by disruption of mitochondrial metabolism. Indeed, the decrease in the NAD:NADH ratio and the associated slowdown of the TCA cycle that are associated with downregulation of respiration are likely to influence the activity of protein metabolic sensors such as sirtuin deacetylases or 2-KG dependent demethylases that may in turn regulate mTOR activity, either directly or indirectly, as suggested in some other contexts (Perez-Gomez, 2020).
Through non-apoptotic roles of caspases, dying cells can release diffusible mitogens and thus signal to their neighbours and instruct them to proliferate -- a process known as apoptosis-induced proliferation (AIP). Several modes of AIP have been described in various species and tissues characterised by the use of either initiator or effector caspase to drive the signalling mechanism that in turn promotes proliferation. In Drosophila wing or eye discs, the most studied AIP models are based on targeted expression of the pro-apoptotic genes hid or rpr where the proliferation is dependent on the initiator caspases that activate ROS production and JNK activity. In this model, proliferation is also dependent on caspases, ROS production and JNK activation, however it is unique in the way it is triggered and in the way the signalling components are interconnected: (1) it is initiated by downregulation of mitochondrial respiratory complex I and therefore it has a metabolic origin (2) it is orchestrated by consequent activation of the TOR pathway (3) it is dependent on effector caspases and (4) JNK activation is upstream of cell death, not activated by the non-apoptotic roles of caspases. Examples of AIP dependent on effector caspases have been described in Drosophila postmitotic cells in the eye disc and in mammalian cells after irradiation induced cell damage but the signalling components involved and their regulatory relationship also differ from the model used in this study (Perez-Gomez, 2020).
It is important to stress that the high levels of ROS species are not produced in every cell where ND-49-RNAi is induced. Although low levels of ROS may appear as a primary consequence of ND-49 downregulation, the strong ROS signal observed in certain areas of the wing disc occurs downstream of cell death, as blocking apoptosis alongside ND-49-RNAi also eliminates the ROS signal. This is in agreement with ROS generation in other modes of AIP. Although ROS production was described with certain complex I inhibitors it does not happen when other inhibitors are used. Assembly of complex I into supercomplexes with other ETC proteins determines if ROS will be produced or not. In the current model it is obvious that the majority of ROS observed does not originate from the dysfunctional complex I but they result from apoptosis, as blocking apoptosis prevents also ROS formation (Perez-Gomez, 2020).
Taken together, the results highlight the central role of TOR pathway activation during mitochondrial dysfunction. As TOR overactivation gives identical phenotype to complex I downregulation, future studies should investigate if the results may be relevant outside of the mitochondria field, in some of the other contexts involving TOR overactivation, such as many types of cancer, wound healing or aging, with potentially important clinical implications (Perez-Gomez, 2020).
One of the most important but less understood step of epithelial tumourigenesis occurs when cells acquire the ability to leave their epithelial compartment. This phenomenon, described as basal epithelial cell extrusion (basal extrusion), represents the first step of tumour invasion. However, due to lack of adequate in vivo model, implication of emblematic signalling pathways such as Ras/Mitogen-Activated Protein Kinase (MAPK) and phosphoinositide 3 kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signalling pathways, is scarcely described in this phenomenon. This paper reports a unique model of basal extrusion in the Drosophila accessory gland. There, it was demonstrated that both Ras/MAPK and PI3K/AKT/mTOR pathways are necessary for basal extrusion. Furthermore, as in prostate cancer, this study shows that these pathways are co-activated. This occurs through set up of Epidermal Growth Factor Receptor (EGFR) and Insulin Receptor (InR) dependent autocrine loops, a phenomenon that, considering human data, could be relevant for prostate cancer (Rambur, 2020).
Worldwide, a large majority of cancers originates from epithelial tissues such as lung, breast and prostate1. Despite reinforced prevention, most of the tumours are detected at late stages, and patient care is centred on invasive adenocarcinomas, resistant forms of these carcinomas and metastatic carcinomas. As late stages of cancer progression have been under intense scrutiny in the last decades, the molecular mechanisms associated to such progression are largely described, showing for example the major role of receptor tyrosine kinase (RTK)-dependent signalling pathways in these mechanisms. Typically, for prostate adenocarcinoma, the second most common cancer in men, both PI3K/AKT/mTOR pathway and Ras/MAPK pathways are associated with tumour progression. In the prostate adenocarcinoma, Ras/MAPK and PI3K/AKT/mTOR pathways display activating genetic alterations in more than 40% of primary tumours and in virtually all metastatic prostate tumours, and phosphoproteomic studies confirmed a strong correlation in the activation of these two pathways. Furthermore, pre-clinical mouse models reproducing alteration of either one or the other pathway in the prostate epithelium display tumourigenesis that mimics histopathological features of the human adenocarcinoma. Moreover, advanced tumour progression is obtained when combining alterations in both pathways. These different data emphasise that in one hand, Ras/MAPK or PI3K/AKT/mTOR pathways can initiate prostate tumour development, and in the other hand that these pathways are implicated in late phases of tumour progression. However, they explain neither their respective or combined role in actual adenocarcinoma formation nor the molecular mechanisms that could couple these two pathways to promote this phenomenon in vivo (Rambur, 2020).
Adenocarcinoma formation occurs when pre-invasive epithelial cells acquire the ability to leave their epithelial compartment. This implicates that these cells are able to extrude from the normal epithelium and to cross the basement membrane which is the limit of the epithelial compartment. These phenomena can be described as basal extrusion and are resulting in early invasion, as opposed to late invasion associated to the metastatic process. Due to the difficulty to precisely visualise basal extrusion in animals, mechanistic associated to this phenomenon has been essentially described in cellular models or in developing tissues such as Drosophila imaginal disc of zebrafish embryo, even though the role of P120 catenin in basal extrusion has been shown in a mouse model of pancreatic neoplasia. The role of Ras/MAPK pathway in basal extrusion has only been described through the use of RasV12 as an oncogenic hit, and role of PI3K/AKT/mTOR pathway has never been assessed (Rambur, 2020).
To determine the role of Ras/MAPK and PI3K/AKT/mTOR pathways in basal extrusion and understand the underlying mechanisms that may coordinate their hyperactivation in prostate cancer, this study has developed a new model of in vivo early invasive adenocarcinoma in the Drosophila prostate-like accessory gland. Drosophila is a powerful genetic model where more than 70% of genes implicated in human diseases display orthologs and where Ras/MAPK and PI3K/AKT/mTOR signalling pathways are well conserved. Drosophila has already proven its pertinence as cancer model for brain, lung, and colon. The Drosophila accessory gland is a functional equivalent for the prostate, playing a role in fertility by secreting seminal fluid. Secretions come from a monolayer of epithelial cells that are well differentiated and quiescent at the adult age, and there is no evidence of stem cells in this tissue. Considering that a majority of prostate adenocarcinoma is thought to originate from luminal cells, epithelial cells from the accessory gland represent a valuable model to study the mechanisms of epithelial prostate tumourigenesis (Rambur, 2020).
This study describes this unique model of basal extrusion and tumour formation in the accessory gland that recapitulates most aspects of cancer development. Both Ras/MAPK and PI3K/Akt/TOR pathways are overactivated in the produced tumours, and these pathways cooperate to induce basal extrusion and subsequent tumour formation. Furthermore, the mechanism is described that allows the coactivation of these pathways, which relies on the sequential recruitment of a double autocrine feedback loop dependent on Epidermal (EGF/Spitz) and Insulin-like (IGF/Ilp6) Growth Factors and their respective receptors. Finally, using publicly available data of prostate cancer samples and migration assay in human pre-tumoural prostate epithelial cell line, the possible role of these findings in the actual human pathology is assessed (Rambur, 2020).
To faithfully reproduce what is thought to happen in the earliest stages of tumour formation in patients, a single genetic alteration was produced in few clones of randomly selected and mostly differentiated cells. Furthermore, accessory gland epithelium, shown to be adjacent to a basement membrane, is surrounded by a stromal-like sheet of muscle fibres, and oncogene-induced epithelial cells are able to cross both layers to form external tumours. This recapitulates the phenomenon of basal epithelial cell extrusion, which is thought to be central to cell invasion. Basal extrusion has been described in cell culture, in Drosophila imaginal discs, in zebrafish embryos and in mouse. However, implication of Ras/MAPK and PI3K/AKT/mTOR pathways has never been assessed in this phenomenon, despite the fact that these pathways are among the most deregulated in cancers, and especially in epithelial cancers such as prostate adenocarcinoma. This study shows in a new model of accessory gland tumourigenesis that both pathways are implicated in basal extrusion, indicating that this step demands a particular state of activation for the cell that undergoes this basal extrusion. Furthermore, this finding correlates with the fact that the two considered pathways are already frequently co-deregulated in primary tumours. From these experiments, where oncogene expression is restricted to few cells and intra-tumoural inhibition of the pathways decreases invasion, it is infered that the mechanisms of basal cell extrusion are cell autonomous, as previously shown in cell lines. Indeed, this study shows that this cell-autonomous mechanism relies on the production of two growth factors, and subsequent activation of two autocrine loops. Role of autocrine loops has been hypothetized in late tumourigenesis, as higher levels of growth factors have been found in tumoural tissues, and has been studied in cell models where inhibition of these loops decreases tumourigenic features such as migration or proliferation capacity as their activation have been linked to transformation of various epithelial cells. However, a role of autocrine loops has never been demonstrated for basal extrusion in vivo. If these loops seem implicated in tumour late progression, so could they be more important for early human tumour development. In fact, many strategies have been attempted to treat cancer patients especially by blocking EGF/EGFR autocrine loop. However, for advanced prostate cancer, these strategies have shown poor results, as well for monotherapies as for combined treatments with classical anti-prostate cancer agents. It could be logical if autocrine loops are less implicated in late stages of cancer but more in the capacity for tumour cells to leave the epithelial compartment. In later stages, higher rates of activating mutations in the Ras/MAPK and PI3K/AKT/mTOR pathways could suppress the need for RTK-driven activation. In contrast, in early tumourigenesis, as fewer genetic alterations are present, activation of signalling pathways must rely on different mechanisms. As is shown in the accessory gland, this recruitment could be efficiently done in tumour cells by autocrine production of growth factors, autocrine activation of their RTK and subsequent activation of the pathways necessary for the tumour development. In a human cohort of prostate cancer samples, it was found that EGF is more expressed in primary tumours than either in normal tissue or in metastases. This could correlate with an early requirement for such growth factor in the formation of adenocarcinoma. Contrary to the observations in Drosophila, no early overexpression of IGFs can be detected in human samples. However, in human, EGFR is able to recruit both Ras/MAPK and PI3K/AKT/mTOR pathway, and EGF overexpression could drive their activation and act in the same way as EGF/Spitz and IGF/Ilp6 in Drosophila (Rambur, 2020).
To study early phases of tumourigenesis remains difficult in vivo, especially for epithelial cells that can develop into benign tumours still in the epithelial compartment such as benign prostatic intraepithelial neoplasia, or into adenocarcinoma that are characterised by an expansion out of the epithelial compartment. The model developed in the Drosophila accessory gland represents a unique in vivo model to explore basal extrusion and early invasion. Two major pathways of cancer progression are implicated in this basal extrusion, and these two pathways are co-recruited by autocrine loops. Further investigation will be necessary to test whether other pathways implicated in late tumourigenesis are important in this phenomenon. Furthermore, it will also be important to determine which genes are activated or inhibited by these pathways and which mechanisms are recruited to promote the actual extrusion (Rambur, 2020).
Target of rapamycin complex 1 (TORC1) is a master regulator of cell metabolism, and its dysregulation has been linked to an array of pathologies, including cancer and age-related diseases. Nprl3, a component of GTPase-activating protein towards Rags complex 1 (GATOR1), inhibits TORC1 activity under nutrient scarcity status. The nprl3 mutant exhibits some metabolic defects due to hyper TORC1 activity in Drosophila. Royal jelly (RJ) is a honeybee-secreted product and plays an essential role in caste differentiation that requires TORC1 activity. RJ is also used as a health-benefit food for its potential roles on antioxidant and anti-aging. In this study, nprl3-mutant flies were used to measure the effect of RJ on metabolic modulation. Interestingly, RJ feeding significantly increased survival and decreased TORC1 activity in the nprl3 mutant. RJ feeding also ameliorated the abnormal reactive oxygen species (ROS) levels and energy status in the nprl3 mutant. The proteins in RJ were characterized to be the essential components in increasing nprl3 mutant viability. These findings suggest that RJ modulates some metabolic defects associated with elevated TORC1 activity and that the nprl3-mutant fly might be a useful tool for investigating the bioactive components of RJ in vivo (Cheng, 2020).
The Myostatin/Activin branch of the TGF-β superfamily acts as a negative regulator of vertebrate skeletal muscle size, in part, through downregulation of insulin/insulin-like growth factor 1 (IGF-1) signaling. Surprisingly, recent studies in Drosophila indicate that motoneuron-derived Activin signaling acts as a positive regulator of muscle size. This study demonstrates that Drosophila Activin signaling promotes the growth of muscle cells along all three axes: width, thickness and length. Activin signaling positively regulates the insulin receptor (InR)/TORC1 pathway and the level of Myosin heavy chain (Mhc), an essential sarcomeric protein, via increased Pdk1 and Akt1 expression. Enhancing InR/TORC1 signaling in the muscle of Activin pathway mutants restores Mhc levels close to those of the wild type, but only increases muscle width. In contrast, hyperactivation of the Activin pathway in muscles increases overall larval body and muscle fiber length, even when Mhc levels are lowered by suppression of TORC1. Together, these results indicate that the Drosophila Activin pathway regulates larval muscle geometry and body size via promoting InR/TORC1-dependent Mhc production and the differential assembly of sarcomeric components into either pre-existing or new sarcomeric units depending on the balance of InR/TORC1 and Activin signals (Kim, 2021).
Insect salivary glands have been shown to function in pupal attachment and food lubrication by secreting factors into the lumen via an exocrine way. This study found in Drosophila that a salivary gland-derived secreted factor (Sgsf) peptide regulates systemic growth via an endocrine way. Sgsf is specifically expressed in salivary glands and secreted into the hemolymph. Sgsf knockout or salivary gland-specific Sgsf knockdown decrease the size of both the body and organs, phenocopying the effects of genetic ablation of salivary glands, while salivary gland-specific Sgsf overexpression increases their size. Sgsf promotes systemic growth by modulating the secretion of the insulin-like peptide Dilp2 from the brain insulin-producing cells (IPCs) and affecting mechanistic target of rapamycin (mTOR) signaling in the fat body. Altogether, this study demonstrates that Sgsf mediates the roles of salivary glands in Drosophila systemic growth, establishing an endocrine function of salivary glands (Li, 2022).
This study shows that the dietary lipid cholesterol, which is required as a component of cell membranes and as a substrate for steroid biosynthesis, also governs body growth and maturation in Drosophila via promoting the expression and release of insulin-like peptides. This nutritional input acts via the nutrient sensor TOR, which is regulated by the Niemann-Pick-type-C 1 (Npc1) cholesterol transporter, in the glia of the blood-brain barrier and cells of the adipose tissue to remotely drive systemic insulin signaling and body growth. Furthermore, increasing intracellular cholesterol levels in the steroid-producing prothoracic gland strongly promotes endoreduplication, leading to an accelerated attainment of a nutritional checkpoint that normally ensures that animals do not initiate maturation prematurely. These findings, therefore, show that a Npc1-TOR signaling system couples the sensing of the lipid cholesterol with cellular and systemic growth control and maturational timing, which may help explain both the link between cholesterol and cancer as well as the connection between body fat (obesity) and early puberty (Texada, 2022).
Animal growth and development depend upon nutrient availability. Therefore, specialized cells and tissues have arisen that sense nutritional inputs and adjust growth and developmental programs via systemic hormonal pathways. In most eumetazoans, these include the conserved insulin-like peptide and steroid-hormone signaling systems. These become dysfunctional when nutrient levels exceed their physiologically normal range. Overloading of the insulin system leads to obesity, metabolic syndrome, insulin resistance, and other pathophysiologies, and overnutrition also leads to precocious puberty associated with childhood obesity (Texada, 2022).
The early life of many animals is a nonreproductive stage of rapid growth, terminated at some nutritional threshold that signals readiness to become a reproductively fit adult. In animals as diverse as humans and insects, this transition is driven by steroid hormones - gonadal steroids including testosterone and estrogen trigger mammalian puberty, and insect metamorphosis is initiated by ecdysone, produced in the prothoracic gland (PG). Similar neuroendocrine cascades regulate insect and mammalian steroidogenesis, including the orthologous neuropeptides Allatostatin A/Kisspeptin and Corazonin/gonadotropin-releasing hormone (GnRH) as well as analogous steroid-feedback circuits. These axes are clearly linked to the metabolic state of the animal, including attainment of a certain critical size. However, the mechanisms of body-size estimation and the effects of nutritional status are not completely understood. Recent work in Drosophila suggests that progression to adulthood is gated by a checkpoint system monitoring tissue growth and nutritional status. When animals reach a 'critical weight' (CW), they become committed to completing their development and maturation, irrespective of further nutritional inputs, whereas animals starved before this checkpoint is satisfied halt their progression to adulthood. This suggests that the CW checkpoint assesses the animal's nutritional state, but the specific nutrients required, and the mechanisms by which their levels are sensed, are incompletely defined (Texada, 2022).
In Drosophila, nutritional input drives growth and maturation through the insulin pathway. Nutrient intake, of amino acids in particular, is sensed via the fat body (analogous to mammalian adipose tissue) and the glia of the blood-brain barrier (BBB). These tissues release factors that regulate the expression and release of Drosophila insulin-like peptides (ILPs) from the insulin-producing cells (IPCs) within the brain, which share functional and developmental homologies with mammalian pancreatic beta cells. These ILPs promote systemic growth through the conserved insulin-receptor/PI3K/Akt pathway. Insulin also promotes PG ecdysone production, linking nutrition directly to developmental progression (Texada, 2022).
Human puberty-triggering CW appears to be linked to body-fat stores, which may explain the link between childhood obesity and early puberty. Despite this, the mechanism by which adiposity affects puberty initiation is unclear. Furthermore, the role of cholesterol has not been considered, even though adipose tissue is a major cholesterol storage depot, especially in obesity. Sterols such as cholesterol have membrane-structural functions but also play important signaling roles, and sterols are required as substrates for steroid-hormone production. Insects, including Drosophila, have lost the ability to synthesize sterols de novo and thus must acquire them through feeding. Mammals are cholesterol prototrophs, but most intracellular cholesterol still comes from low-density-lipoprotein (LDL)-mediated cellular uptake of dietary cholesterol. In both taxa, consumed sterols are transported in the circulatory system bound within lipoprotein particles (LPPs such as mammalian LDL/HDL), and target tissues take them up through a variety of mechanisms including receptor-mediated endocytosis. LPP-bound sterols are extracted in the lysosome and inserted into the lysosomal membrane by membrane-integral transport proteins. The primary such protein, Niemann-Pick-type-C 1 (Npc1), underlies the Niemann-Pick type C lysosomal storage disorder; without Npc1 function, cholesterol accumulates in endosomal-lysosomal compartments, leading to increased intracellular cholesterol signaling. Thus, Npc1 seems to be part of a mechanism by which cells regulate cholesterol signaling (Texada, 2022).
This study set out to determine whether, and the routes by which, cholesterol might regulate Drosophila larval growth. The findings show that dietary cholesterol dose-responsively promotes growth and accelerates development by increasing insulin signaling. Cholesterol sensing is mediated by the target of rapamycin (TOR) pathway in the cells of the fat body and the glia of the BBB, which remotely induce the expression and release of ILPs from the IPCs. Enhancing cholesterol signaling in the PG also promotes TOR activity, drives endoreduplication, and leads to premature attainment of the CW checkpoint. Thus, dietary cholesterol accelerates growth through insulin signaling and leads to early maturation through effects on steroidogenesis, effects which are mediated by promoting TOR activity in sensing tissues (Texada, 2022).
Nutrition is one of the most important influences on developmental growth and maturation. Malnutrition or disease can impair growth and delay puberty, whereas obese children enter puberty early. Similarly, Drosophila larvae exposed to poor nutrition, tissue damage, or inflammation delay their development, whereas rich conditions promote rapid growth and maturation. These environmental factors are coupled to the appropriate gating of steroid production via internal checkpoints, one of which is a nutrition-dependent CW required to initiate the maturation process. This suggests that signals reflecting nutritional state and body-fat storage play a key role in activating the neuroendocrine pathways that trigger puberty. Although studies suggest that the adipokine leptin may be involved, the mechanisms linking body fat to puberty are poorly defined, and the potential involvement of lifestyles associated with excessive accumulation of cholesterol, one of the most important lipids, has not been considered. In humans, white adipose tissue is the main site of cholesterol storage and can contain over half the body's total cholesterol in obesity. The results show that dietary cholesterol intake promotes systemic body growth through insulin-dependent pathways and that animals raised on high dietary cholesterol initiate maturation earlier. Cholesterol is sensed through an Npc1-regulated TOR-mediated mechanism in the fat body and the glial cells of the BBB, which relay information to the IPCs within the brain to promote insulin expression and release, thus coupling growth and maturation with cholesterol status (Texada, 2022).
Insect CW likely evolved as a mechanism ensuring that maturation will not occur unless the animal has accumulated adequate nutrient stores to survive the nonfeeding metamorphosis period and has completed sufficient growth to produce an adult of proper size and thus of maximal fitness. Likewise, the link between body fat and maturation in humans probably ensures adequate stores of fat before maturation onset to support pregnancy and reproductive success. In Drosophila, insulin signaling plays a critical role in coordinating steroidogenesis with nutritional conditions. Insulin acts upon the PG and induces a small ecdysone peak early in L3 that is correlated with CW attainment. In combination with nutrition-related signaling mediated via insulin, nutrient availability is also assessed directly in the PG and is coupled to irreversible endoreduplication that permits ecdysone production at CW. the current findings show that accumulation of cholesterol in the PG, induced by the loss of Npc1a, drives a remarkable TOR-dependent increase in endoreduplication and leads to inappropriate attainment of CW. Taken together, These findings indicate that cholesterol sensed by the BBB glia and the fat body promotes growth through insulin signaling and that cholesterol sensed by the PG accelerates maturation through ecdysone signaling (Texada, 2022).
Loss of Npc1 function in humans leads to the Niemann-Pick lysosomal storage disorder, marked by intracellular cholesterol accumulation. Although neurodegeneration is the hallmark of NPC disease, including in a Drosophila model, alterations in glial, adipose, hepatic, and endocrine systems are also components of NPC syndrome. In humans, Npc1 itself is strongly expressed in glia and in adipose tissues, especially in obese individuals, and variants in Npc1 are associated with obesity, type-2 diabetes, and hepatic lipid dysfunction. These findings link glial and adipose-tissue cholesterol sensing through Npc1 to systemic growth and metabolic control through effects on insulin signaling. It was also found that intracellular cholesterol accumulation driven by Npc1 loss leads to hyperactivation of TOR that drives increases in DNA replication and cell growth. TOR activity is frequently upregulated in cancer, and the results therefore provide mechanistic insight for understanding the emerging link between cholesterol and a range of cancers (Texada, 2022).
As the coupling of nutrition with growth and maturation is ancient and highly conserved, this work provides a foundation for understanding how cholesterol is coupled to developmental growth and maturation initiation in humans. These findings link a high concentration of this particular lipid in adipose tissues to the neuroendocrine initiation of maturation, which may explain the critical link between obesity (body fat) and early puberty (Texada, 2022).
Precise organ size control is fundamental for all metazoans, but how organ size is controlled in a three-dimensional (3D) way remains largely unexplored at the molecular level. This study screened and identified Drosophila Ptp61F as a pivotal regulator of organ size that integrates the Hippo pathway, TOR pathway, and actomyosin machinery. Pathologically, Ptp61F loss synergizes with Ras(V12) to induce tumorigenesis. Physiologically, Ptp61F depletion increases body size and drives neoplastic intestinal tumor formation and stem cell proliferation. Ptp61F also regulates cell contractility and myosin activation and controls 3D cell shape by reducing cell height and horizontal cell size. Mechanistically, Ptp61F forms a complex with Expanded (Ex) and increases endosomal localization of Ex and Yki. Furthermore, it was demonstrated that PTPN2, the conserved human ortholog of Ptp61F, can functionally substitute for Ptp61F in Drosophila. This work defines Ptp61F as an essential determinant that controls 3D organ size under both physiological and pathological conditions (Liu, 2022).
Along with differences in life histories, metazoans have also evolved vast differences in cellularity, involving changes in the molecular pathways controlling the cell cycle. The extent to which the signalling network systemically determines cellular composition throughout the body and whether tissue cellularity is organized locally to match tissue-specific functions are unclear. This study cultured genetic lines of Drosophila melanogaster on food with and without rapamycin to manipulate the activity of target of rapamycin (TOR)/insulin pathways and evaluate cell-size changes in five types of adult cells: wing and leg epidermal cells, ommatidial cells, indirect flight muscle cells and Malpighian tubule epithelial cells. Rapamycin blocks TOR multiprotein complex 1, reducing cell growth, but this effect has been studied in single cell types. As adults, rapamycin-treated flies had smaller bodies and consistently smaller cells in all tissues. Regardless, females eclosed with larger bodies and larger cells in all tissues than males. Thus, differences in TOR activity and sex were associated with the orchestration of cell size throughout the body, leading to differences in body size. It is postulated that the activity of TOR/insulin pathways and their effects on cellularity should be considered when investigating the origin of ecological and evolutionary patterns in life histories (Szlachcic, 2023).
Oncogenes often promote cell death as well as proliferation. How oncogenes drive these diametrically opposed phenomena remains to be solved. A key question is whether cell death occurs as a response to aberrant proliferation signals or through a proliferation-independent mechanism. This study revealed that Src, the first identified oncogene, simultaneously drives cell proliferation and death in an obligatorily coupled manner through parallel MAPK pathways. The two MAPK pathways diverge from a lynchpin protein Slpr. A MAPK p38 drives proliferation whereas another MAPK JNK drives apoptosis independently of proliferation signals. Src-p38-induced proliferation is regulated by methionine-mediated Tor signaling. Reduction of dietary methionine uncouples the obligatory coupling of cell proliferation and death, suppressing tumorigenesis and tumor-induced lethality. These findings provide an insight into how cells evolved to have a fail-safe mechanism that thwarts tumorigenesis by the oncogene Src. This study also exemplifies a diet-based approach to circumvent oncogenesis by exploiting the fail-safe mechanism (Nishida, 2021).
This study elucidated the mechanism by which Src drives cell proliferation and cell death in an obligatory coupled manner. The obligation is mediated by coupling of two MAPK pathways diverging from the lynchpin protein Slpr. Downstream of Slpr, JNK activates cell death signaling, while p38 activates cell proliferation in a methionine-Tor dependent manner. Src can potentially regulate Tor signaling through both p38-dependent and -independent mechanisms. This work provides several new insights discussed below (Nishida, 2021).
First, the findings that Slpr mediates Src signaling provide a new molecular insight into regulation of Src signaling. Drosophila Src has been known to regulate various signaling pathways, including Notch, MAPKs, Jak-Stat, EGF, Wnt, and Hippo signaling, but Slpr has not previously been implicated in Src signaling. Especially, the mechanism behind Src-mediated JNK activation was elusive in spite of its biological importance in various contexts. Slpr fills in the gap between Src and JNK. In hindsight, it may seem sensible that Slpr, a JNKKK, could link Src and JNK. However, previous studies proposed that ubiquitin E2 complex Bendless and F-actin cytoskeleton mediate Src-JNK signaling. Thus, it was unclear until now whether a MAPKKK is necessary for Src-mediated activation of JNK. Furthermore, there are five Drosophila JNKKKs, including dTAK1, Mekk1, Ask1, Wnd, and Slpr, each of which functions uniquely in a context-dependent manner. In an initial RNAi screening that identified Slpr as a Src effector, other MAPKKKs were not identified. Thus, identification of Slpr as a linker between Src and JNK provides a new insight. An urging, next question is how Src regulates Slpr. It is speculated that the components that are considered as Src downstream and/or Slpr; upstream, such as Dok, Shark, and Misshapen, may mediate the signal transduction between them. Interestingly, it was also found that Slpr inhibition suppresses the phenotype of CA Ras overexpression, which, similar to Src, simultaneously induces apoptosis and proliferation. This suggests that Slpr could function as a lynchpin hub that integrates inputs from multiple oncogenes (Nishida, 2021).
This study exclusively focused on cell autonomous signaling induced by Src. But it was noticed that Src elicits non-cell autonomous activation of MAPKs, cell death, and proliferation, This is reminiscent of the non-cell autonomous activation of Yorkie by Src. It will be interesting to elucidate how non-cell autonomous signaling is regulated by Src activation in a future study (Nishida, 2021).
Second, although Src was known to induce apoptosis as well as cell proliferation, how Src accomplishes this was unclear. This study elucidated that, diverging from Slpr, p38 accelerates cell proliferation and that JNK induces cell death. This is an obligatory coupling of proliferation and death, likely being accomplished through evolution as an imperative mechanism to prevent tumorigenesis by a single oncogene activation. This type of fail-safe mechanism to prevent facile transformation was previously suggested in a context of Myc oncogene. It is proposed that, although each oncogene should have its unique fail-safe mechanism, the concept of the intrinsic fail-safe mechanism to prevent oncogenesis by a single oncogene is general (Nishida, 2021).
Third, from a therapeutic perspective, the observation that methionine strongly regulates Src-mediated overgrowth is intriguing. Tumor growth in vitro is metabolically regulated by nutrition and dietary manipulation of serine, glycine, histidine, asparagine, cysteine, or methionine could clinically modulate cancer outcome. Notably, in the physiological in vivo condition, only subtraction of methionine from diet enhances organismal survival over Src-mediated oncogenic stress. Methionine has been studied in contexts of life span, metabolic health, and cancer together with other amino acids, but the molecular mechanisms behind methionine-mediated cellular and organismal physiology were often unclear. This study demonstrates that methionine regulates Tor activation, which controls cell proliferation induced by Src-p38 signaling (Nishida, 2021).
This study also found that the methionine concentration in the hemolymph is lower in flies that bear tumors in the wing disc. This is reminiscent of the clinical condition where tumor affects the amino acid profiles in the blood. Of note, local glutamine is known to be consumed in the tumor environment, but at least reduction of glutamine in the hemolymph of the flies bearing tumors was not observed. It is presumed that Src-induced increase of methionine uptake in the Src tumor is at least partly responsible for the Src tumor-induced hypomethioninemia, although other tissues may also contribute to it as the case with the fat body during wing disc repair (Nishida, 2021).
Regarding a cross-talk between Src signaling and nutrition-mediated Tor activation, this study found that there are multiple cross-talk points. Src regulates methionine uptake and methionine flux in a p38-independent manner, both of which can potentially feed into Tor activation. Then, a question is how Src-p38 regulates Tor signaling, since Src-p38 clearly activates Tor signaling. Although p38 is known to regulate Tor, its exact molecular mechanism remains unclear. Using the previously published RNAseq data on Src tumor in the wing disc, expression levels of potential Tor regulators were surveyed and genes were selected that are affected by Src expression, including amino acid transporters and GATOR complexes. GATOR complexes regulate Tor through Rag GTPases. This study examined whether their expression is regulated by Src in a p38-dependent manner using RT-qPCR. Among the amino acid transporters and GATOR complex components examined, only pathetic (path), an SLC36 amino acid transporter that can transport multiple amino acids, was significantly induced by Src in a p38-dependent manner. Since Path can mediate amino acids-mediated Tor activation, it is speculated that Src-p38 could regulate Tor potentially through Path-mediated uptake of non-methionine amino acids (Nishida, 2021).
These findings have significant implications in the field of cancer therapeutics. As described in Introduction, SFK inhibitors have been clinically unsuccessful in spite of SFKs' contribution to tumorigenesis and metastasis. It is expected that the new insights this study provides on the Src tumorigenesis may help pave the way to cancer treatment. Furthermore, the data imply that nutritional state and tumorigenesis are closely linked. It is speculated that, in case of tumors with a high SFK activity, manipulation of dietary methionine may have a clinical benefit (Nishida, 2021).
Mechanistic Target of Rapamycin Complex 1 (mTORC1) is a central regulator of cell growth and metabolism that senses and integrates nutritional and environmental cues with cellular responses. Recent studies have revealed critical roles of mTORC1 in RNA biogenesis and processing. This study finds that the m(6)A methyltransferase complex (MTC) is a downstream effector of mTORC1 during autophagy in Drosophila and human cells. Furthermore, the Chaperonin Containing Tailless complex polypeptide 1 (CCT) complex, which facilitates protein folding, acts as a link between mTORC1 and MTC. The mTORC1 activates the chaperonin CCT complex to stabilize MTC, thereby increasing m(6)A levels on the messenger RNAs encoding autophagy-related genes, leading to their degradation and suppression of autophagy. Altogether, this study reveals an evolutionarily conserved mechanism linking mTORC1 signaling with m(6)A RNA methylation and demonstrates their roles in suppressing autophagy (Tang, 2021).
mTORC1, an evolutionarily conserved serine/threonine kinase, is a master regulator of cell growth, metabolism, and proliferation coupling different nutritional and environmental cues, including growth factors, energy levels, cellular stress, and amino acids, with metabolic programs. For example, insulin activates PI3K/AKT and inhibits the Tuberous Sclerosis Complex (TSC) 1/2, a negative regulator of mTORC1, thus promoting mTORC1 activation. Activated mTORC1 then phosphorylates multiple downstream effectors that control a wide range of anabolic and catabolic processes. Phosphorylation of the ribosomal S6 kinase 1 (S6K1) and eIF4E-binding protein 1 (4E-BP1) by mTORC1 promotes protein translation and enhances cell growth and proliferation. Moreover, autophagy, an intracellular degradation system that delivers cytoplasmic components to lysosomes, is inhibited by mTORC1 through phosphorylation of Atg13 that, in turn, inhibits ULK1 kinase activity (Tang, 2021).
Recent studies have highlighted a role for mTORC1 in regulating RNA metabolism. Through the phosphorylation of RNA metabolic proteins, mTORC1 modulates various RNA biogenesis and processing events. Phosphorylation of the SR protein kinase SRPK2 by S6K1 promotes its transport into the nucleus where it activates SR proteins and induces splicing of lipogenic pre-messenger RNAs (pre-mRNAs) for de novo synthesis of fatty acids and cholesterol, suggesting that SRPK2 is a critical mediator of mTORC1-dependent lipogenesis. In addition, mTORC1 regulates alternative splicing and polyadenylation of autophagic and metabolic genes to control autophagy, lipid, protein, and energy metabolism through the cleavage and polyadenylation complex. Furthermore, mTORC1 mediates phosphorylation of the decapping enzyme Dcp2. Phosphorylated Dcp2 associates with RNA helicase RCK family members and binds to transcripts of Autophagy-related genes (Atg) to degrade them, thereby suppressing autophagy. Altogether, these studies suggest an essential role for mTORC1 in controlling RNA biogenesis and processing, revealing a major function for mTORC1 in the regulation of protein diversity and in reshaping cellular metabolism and autophagy (Tang, 2021).
N6-methyl-adenosine (m6A) is one of the most abundant chemical modifications in eukaryotic mRNA, which is preferentially enriched in 3' UTRs and around stop codons. m6A modification affects almost all aspects of mRNA metabolism, such as splicing, translation, and stability, and plays essential roles in a wide range of cellular processes, including Drosophila sex determination and metabolism. The m6A methyltransferase complex (MTC) catalyzes m6A formation and is composed of the methyltransferase-like protein 3 (METTL3), the methyltransferase-like protein 14 (METTL14), WTAP (the ortholog of Drosophila Fl(2)d), and RBM15/RBM15B (the ortholog of Drosophila Nito). Although METTL3 is the only catalytic component of the MTC, its interaction with METTL14 is necessary for RNA substrate recognition and efficient m6A deposition. WTAP stabilizes the interaction between the two METTL proteins, and RBM15/RBM15B have been proposed to recruit the MTC to its target transcripts (Tang, 2021).
Using autophagy as a readout of mTORC1 signaling in Drosophila, the MTC was identified as a downstream effector of mTORC1 signaling. From the analysis of high-confidence Drosophila and human MTC proteomic data, the Chaperonin Containing Tailless complex polypeptide 1 (CCT) complex was identified as an MTC interactor that mediates the effects of mTORC1 on m6A modification and autophagy. In mammalian cells, it was also found that the CCT complex plays critical roles in the regulation of MTC protein stability and m6A RNA modification, suggesting that the mTORC1-CCT-MTC axis is conserved from Drosophila to mammals. These studies thus unveil a mechanism linking mTORC1 signaling and the chaperonin CCT complex to RNA methylation and also uncover a layer of mTORC1 regulation of autophagy (Tang, 2021).
This study has demonstrate that the MTC acts as a downstream effector of mTORC1 to regulate m6A RNA methylation of Atg transcripts, inducing their degradation and thus suppressing autophagy. Furthermore, the CCT complex was identified as a link between mTORC1 and MTC. CCT downstream of mTORC1 signaling can stabilize METTL3 and METTL14 to up-regulate m6A levels and inhibit autophagy. Accordingly, depletion of either mTORC1, CCT, METTL3, or METTL14 compromises m6A RNA methylation and promotes autophagy. Importantly, the role of mTORC1-CCT-MTC signaling in regulating autophagy is conserved from Drosophila to mammals. Thus, this study discovered a function of mTORC1 in regulating m6A RNA methylation during autophagy (Tang, 2021).
mTORC1 inhibition suppresses protein translation but also affects gene expression at different levels. This study identified an epitranscriptomic mechanism by which mTORC1 activates m6A RNA methylation to promote Atg mRNA turnover and inhibits autophagy. This m6A-mediated mRNA degradation represents a layer of gene regulation by mTORC1. Moreover, as mTORC1 activity regulates global m6A levels, it is likely that the MTC also mediates additional physiological functions of mTORC1. It is noted that depletion of METTL3, METTL14, or CCT8 cannot fully rescue TSC1-induced effects. Although these results could be caused by partial RNAi knockdown, they may also indicate that other pathways contribute to mTORC1 regulation of autophagy. Indeed, studies have reported that mTORC1 suppresses autophagy through modulation of transcription factors, RNA-processing complexes, and mRNA degradation machinery, further highlighting that mTORC1 utilizes multiple RNA biogenesis processes to control autophagy (Tang, 2021).
The catalytic core components of the MTC, METTL3/METTL14, have a substrate sequence specificity for a DRA*CH motif (D = G/A/U, R = G/A, A* = methylated adenosine, H = A/U/C). However, only a subset of consensus sites across the mRNA transcriptome are methylated. Thus, it has been speculated that other factors in the MTC specify METTL3/METTL14 methylation patterns. Proteomic results combined with biochemical validation in both Drosophila and mammalian cells identified multiple splicing factors that interact with known MTC components. Future work will be needed to confirm whether these factors are directly involved in the regulation of m6A methylation and how they coordinate with the m6A machinery to affect RNA processing. It will also be interesting to investigate whether mTORC1 controls other regulators of RNA m6A methylation, in addition to METTL3 and METTL14. Moreover, the proteomics data revealed that multiple components of E3 ubiquitin ligase complex interact with MTC, suggesting that they may be involved in ubiquitination of MTC. Ubiquitination of METTL3 has also been observed, but its function and related E3 ubiquitin ligases remain unclear (Tang, 2021).
Previous genetic analyses showed that the CCT complex functions downstream of mTORC1 and that mTORC1 positively regulates the transcriptional levels of the CCT complex (Kim, 2019). Another study identified CCT2 as a substrate of S6 kinase, a downstream effector of mTOR, in mammalian cells, suggesting that both transcriptional and posttranslational regulations contribute to CCT complex activation by mTORC1. However, the phosphorylation site (Ser-260) of mammalian CCT2 is not conserved in Drosophila and how this phosphorylation modulates CCT function is not clear. Multiple phosphorylation sites have been detected in CCT components. Interestingly, a previous study showed that CCT8 was phosphorylated following insulin stimulation (Vinayagam, 2016), suggesting that other phosphorylation sites are involved in mTORC1-regulated CCT activation. Future studies are needed to comprehensively map the phosphorylation sites on CCT components and investigate their physiological roles (Tang, 2021).
The CCT complex is a highly conserved complex that assists the folding of about 10% of the eukaryotic proteome. The interactions of the CCT complex with METTL3 and METTL14 were observed in a previous study using AP/MS in human cells. Consistently, genetic and biochemical data from this study further confirmed their interactions and characterized the functions of CCT in stabilizing METTL3 and METTL14 and controlling m6A RNA methylation. These findings thus further expand the impact of the CCT complex on RNA metabolism (Tang, 2021).
Multiple studies have reported that CCT complex protein levels dramatically increase in autophagy mutants, proposing that CCT is one of the substrates of autophagy. Future studies will be needed to test whether autophagy is able to degrade the CCT complex and whether autophagy feedback inhibits CCT (Tang, 2021).
Loss-of-function and gain-of-function genetic perturbations provide valuable insights into gene function. In Drosophila cells, while genome-wide loss-of-function screens have been extensively used to reveal mechanisms of a variety of biological processes, approaches for performing genome-wide gain-of-function screens are still lacking. This study describe a pooled CRISPR activation (CRISPRa) screening platform in Drosophila cells and apply this method to both focused and genome-wide screens to identify rapamycin resistance genes. The screens identified three genes as novel rapamycin resistance genes: a member of the SLC16 family of monocarboxylate transporters (CG8468), a member of the lipocalin protein family (CG5399), and a zinc finger C2H2 transcription factor (CG9932). Mechanistically, it was demonstrated that CG5399 overexpression activates the RTK-Akt-mTOR signaling pathway and that activation of insulin receptor (InR) by CG5399 requires cholesterol and clathrin-coated pits at the cell membrane. This study establishes a novel platform for functional genetic studies in Drosophila cells (Xia, 2023).
Expansion of G4C2 hexanucleotide repeats in the chromosome 9 ORF 72 (C9ORF72) gene is the most common genetic cause of amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (C9-ALS/FTD). Dipeptide repeats generated by unconventional translation, especially the R-containing poly(GR), have been implicated in C9-ALS/FTD pathogenesis. Mutations in other genes, including TAR DNA-binding protein 43 KD (TDP-43), fused in sarcoma (FUS), and valosin-containing protein, have also been linked to ALS/FTD, and upregulation of amyloid precursor protein (APP) is observed at the early stage of ALS and FTD. Fundamental questions remain as to the relationships between these ALS/FTD genes and whether they converge on similar cellular pathways. In this study, using biochemical, cell biological, and genetic analyses in Drosophila disease models, patient-derived fibroblasts, and mammalian cell culture, it was shown that mechanistic target of rapamycin complex 2 (mTORC2)/AKT signaling is activated by APP, TDP-43, and FUS and that mTORC2/AKT and its downstream target valosin-containing protein mediate the effect of APP, TDP-43, and FUS on the quality control of C9-ALS/FTD-associated poly(GR) translation. This study also found that poly(GR) expression results in reduction of global translation and that the coexpression of APP, TDP-43, and FUS results in further reduction of global translation, presumably through the GCN2/eIF2α-integrated stress response pathway. Together, these results implicate mTORC2/AKT signaling and GCN2/eIF2α-integrated stress response as common signaling pathways underlying ALS/FTD pathogenesis (Li, 2023).
Focal epilepsy accounts for 60% of all forms of epilepsy, but the pathogenic mechanism is not well understood. In this study, three novel mutations in NPRL3 (nitrogen permease regulator-like 3), c.937_945del, c.1514dupC and 6,706-bp genomic DNA (gDNA) deletion, were identified in three families with focal epilepsy by linkage analysis, whole exome sequencing (WES) and Sanger sequencing. NPRL3 protein is a component of the GATOR1 complex, a major inhibitor of mTOR signaling. These mutations led to truncation of the NPRL3 protein and hampered the binding between NPRL3 and DEPDC5, which is another component of the GATOR1 complex. Consequently, the mutant proteins enhanced mTOR signaling in cultured cells, possibly due to impaired inhibition of mTORC1 by GATOR1. Knockdown of nprl3 in Drosophila resulted in epilepsy-like behavior and abnormal synaptic development. Taken together, these findings expand the genotypic spectrum of NPRL3-associated focal epilepsy and provide further insight into how NPRL3 mutations lead to epilepsy (Du, 2023).
Tumor cells have an increased demand for nutrients to sustain their growth, but how these increased metabolic needs are ensured or how this influences tumor formation and progression remains unclear. To unravel tumor metabolic dependencies, particularly from extracellular metabolites, this study has analyzed the role of plasma membrane metabolic transporters in Drosophila brain tumors. Using a well-established neural stem cell-derived tumor model, caused by brat knockdown, this study has found that 13 plasma membrane metabolic transporters, including amino acid, carbohydrate and monocarboxylate transporters, are upregulated in tumors and are required for tumor growth. CD98hc, and several of the light chains with which it can form heterodimeric amino acid transporters, were identified as crucial players in brat RNAi (brat (IR)) tumor progression. Knockdown of these components of CD98 heterodimers caused a dramatic reduction in tumor growth. The data also reveal that the oncogene dMyc is required and sufficient for the upregulation of CD98 transporter subunits in these tumors. Furthermore, tumor-upregulated dmyc and CD98 transporters orchestrate the overactivation of the growth-promoting signaling pathway TOR, forming a core growth regulatory network to support brat (IR) tumor progression. These findings highlight the important link between oncogenes, metabolism, and signaling pathways in the regulation of tumor growth and allow for a better understanding of the mechanisms necessary for tumor progression (Rebelo, 2023).
PDGF/VEGF ligands regulate a plethora of biological processes in multicellular organisms via autocrine, paracrine and endocrine mechanisms. This study investigated organ-specific metabolic roles of Drosophila PDGF/VEGF-like factors (Pvfs). Genetic approaches and single-nuclei sequencing were combined to demonstrate that muscle-derived Pvf1 signals to the Drosophila hepatocyte-like cells/oenocytes to suppress lipid synthesis by activating the Pi3K/Akt1/TOR signaling cascade in the oenocytes. Functionally, this signaling axis regulates expansion of adipose tissue lipid stores in newly eclosed flies. Flies emerge after pupation with limited adipose tissue lipid stores and lipid level is progressively accumulated via lipid synthesis. This study found that adult muscle-specific expression of pvf1 increases rapidly during this stage and that muscle-to-oenocyte Pvf1 signaling inhibits expansion of adipose tissue lipid stores as the process reaches completion. These findings provide the first evidence in a metazoan of a PDGF/VEGF ligand acting as a myokine that regulates systemic lipid homeostasis by activating TOR in hepatocyte-like cells (Ghosh, 2020).
The presence in vertebrates of multiple PDGF/VEGF signaling ligands and cognate receptors makes it difficult to assess their roles in inter-organ communication. Additionally, understanding the tissue-specific roles of these molecules, while circumventing the critical role they play in regulating tissue vascularization, is equally challenging in vertebrate models. This study investigated the tissue-specific roles of the ancestral PDGF/VEGF-like factors and the single PDGF/VEGF-receptor in Drosophila in lipid homeostasis. The results demonstrate that in adult flies the PDGF/VEGF like factor, Pvf1, is a muscle-derived signaling molecule (myokine) that suppresses systemic lipid synthesis by signaling to the Drosophila hepatocyte-like cells/oenocytes (Ghosh, 2020).
The Drosophila larval and adult adipose tissues have distinct developmental origins. The larval adipose tissue undergo drastic morphological changes during metamorphosis and dissociate into individual large spherical cells. These free-floating adipose cells persist to the young adult stage where they play a crucial role in protecting the animal from starvation and desiccation. These larval adipose tissue cells are ultimately removed via cell death. Adult-specific adipose tissue cells develop during the pupal stage from yet unknown progenitor cells and have very little lipid stores in newly eclosed flies. Over the period of next 3-5 days the adult adipose tissue builds up its lipid reserves through feeding and de-novo lipid synthesis. However, at the end of the lipid build-up phase, the rate of lipid synthesis must be suppressed to avoid over-loading of the adipose tissue and prevent lipid toxicity. The data suggest that muscle Pvf1 signaling suppresses lipid synthesis at the end of the adult adipose tissue lipid build-up phase. Pvf1 production in the adult muscles peaks around the time when adult adipose tissue lipid stores reach steady state capacity. In turn, muscle-derived Pvf1 suppresses lipid synthesis and lipid incorporation by activating TOR signaling in the oenocytes (Ghosh, 2020).
This study reveals that Pvf1 is abundant in the tubular muscles of the Drosophila leg and abdomen. In these striated muscles, the protein localizes between individual myofibrils and is particularly enriched at the M and Z bands. Drosophila musculature can be broadly categorized into two groups, the fibrillar muscles and the tubular muscles, with distinct morphological and physiological characteristics. Drosophila IFMs of the thorax belong to the fibrillar muscle group and are stretch-activated, oxidative, slow twitch muscles that are similar to vertebrate cardiac muscles. By contrast, Drosophila leg muscles and abdominal muscles belong to the tubular muscle group. These muscles are striated, Ca2+ activated, and glycolytic in nature. The tubular muscles are structurally and functionally closer to vertebrate skeletal muscles. Expression of Pvf1 in the tubular muscles of the Drosophila leg may reflect a potentially conserved role of this molecule as a skeletal-muscle-derived myokine. The fact that most of the myokines in vertebrates were identified in striated skeletal muscles supports this possibility . Moreover, vertebrate VEGF ligands, VEGF-A and VEGF-B, have also been shown to be stored and released from skeletal muscles (Ghosh, 2020).
Interestingly, in vertebrates, the expression and release of VEGF ligands are regulated by muscle activity. In mice, expression of VEGF-B in the skeletal muscles is regulated by PGC1-α, one of the key downstream effectors of muscle activity. Additionally, expression of VEGF-B is upregulated in both mouse and human skeletal muscles in response to muscle activity. Similarly, expression of VEGF-A is induced by muscle contraction. No effect of muscle activity on the expression levels of pvf1 was observed in the Drosophila muscles. Whether muscle activity regulates release of Pvf1 primarily could not be demonstrated due to the difficulty in collecting adequate amounts of hemolymph from the adult males. However, the localization of Pvf1 to the M/Z bands suggests a potential role for muscle activity in Pvf1 release. The M and Z bands of skeletal muscles are important centers for sensing muscle stress and strain. These protein-dense regions of the muscle house a number of proteins that can act as mechano-sensors and mediate signaling events including translocation of selected transcription factors to the nucleus. Pvf1, therefore, is ideally located to be able to sense muscle contraction and be released in response to muscle activity. Further work, contingent on the development of new tools and techniques, will be necessary to measure Pvf1 release into the hemolymph and study the regulation of this release by exercise (Ghosh, 2020).
Previous work has shown that Pvf1 released from gut tumors generated by activation of the oncogene yorkie leads to wasting of Drosophila muscle and adipose tissue (Song, 2019). Adipose tissue wasting in these animals is characterized by increased lipolysis and release of free fatty acids (FFAs) in circulation. However, no role was observed of Pvf signaling in regulating lipolysis in the adipose tissue of healthy well-fed flies without tumors. Loss of PvR signaling in the adipose tissue did not have any effect on lipid content. Additionally, over-expressing Pvf1 in the muscle did not lead to the bloating phenotype commonly seen in cachectic animals with gut tumors. It is concluded that Pvf1 affects wasting of the adipose tissue only in the context of gut tumors and that the effect could involve the following mechanisms: (1) the gut tumor releases pathologically high levels of Pvf1 into circulation leading to ectopic activation of PvR signaling in the adipose tissue, and, that such high levels of Pvf1 are not released by the muscle (even when pvf1 is over-expressed in the muscle); (2) Pvf1 causes adipose tissue wasting in the context of other signals that emanate from the gut tumor that are not available in flies that do not have tumors (Ghosh, 2020).
Only oenocyte-specific loss of PvR signaling phenocopies the obesity phenotype caused by muscle-specific loss of Pvf1, indicating that muscle-Pvf1 primarily signals to the oenocytes to regulate systemic lipid content. Additionally, muscle-specific loss of Pvf1, as well as oenocyte-specific loss of PvR and its downstream effector TOR, leads to an increase in the rate of lipid synthesis. These observations indicate a role for the Drosophila oenocytes in lipid synthesis and lipid accumulation in the adipose tissue. Oenocytes have been implicated in lipid metabolism previously and these cells are known to express a diverse set of lipid metabolizing genes including but not limited to fatty acid synthases, fatty acid desaturases, fatty acid elongases, fatty acid β-oxydation enzymes and lipophorin receptors. Functionally, the oenocytes are proposed to mediate a number of lipid metabolism processes. Oenocytes tend to accumulate lipids during starvation (presumably for the purpose of processing lipids for transport to other organs and generation of ketone bodies) and are necessary for starvation induced mobilization of lipids from the adipose tissue. This role is similar to the function of the liver in clearing FFAs from circulation during starvation for the purpose of ketone body generation, and redistribution of FFAs to other organs by converting them to TAG and packaging into very-low density lipoproteins. However, a [1-14C]-oleate chase assay did not show any effect of oenocyte-specific loss of PvR/TOR signaling on the rate of lipid utilization, indicating that this pathway does not affect oenocyte-dependent lipid mobilization (Ghosh, 2020).
Oenocytes also play a crucial role in the production of very-long-chain fatty acids (VLCFAs) needed for waterproofing of the cuticle (Storelli, 2019). Results of a starvation resistance assay indicate that loss of the muscle-to-oenocyte Pvf1 signaling axis does not affect waterproofing of the adult cuticle. Storelli (2019) recently showed that the lethality observed in traditionally used starvation assays is largely caused by desiccation unless the assay is performed under saturated humidity conditions. Since a starvation assay was performed under 60% relative humidity (i.e. non-saturated levels), it is likely that desiccation played a partial role in causing starvation-induced lethality. Any defects in waterproofing of the adult cuticle would have led to reduced starvation resistance. However, both muscle-specific loss of Pvf1 and oenocyte-specific loss of PvR led to increased starvation resistance suggesting normal waterproofing in these animals. The increased starvation resistance in these animals is likely the result of these animals having higher stored lipid content that helps them to survive longer without food (Ghosh, 2020).
Insect oenocytes were originally believed to be lipid synthesizing cells because they contain wax-like granules. These cells express a large number of lipid-synthesizing genes and the abundance of smooth endoplasmic reticulum further suggest a role for this organ in lipid synthesis and transport. However, evidence for potential involvement of the oenocytes in regulating lipid synthesis and lipid deposition in the adipose tissue has been lacking. The fact that two of the three fatty acid synthases (fasn2 and fasn3) encoded by the Drosophila genome are expressed specifically in adult oenocytes suggests a potential role for these cells in lipid synthesis. The observation that oenocyte-specific loss of PvR and its downstream effector TOR leads to increased lipid synthesis and increased lipid content of the adipose tissue strongly supports this possibility. The data further suggests that involvement of the oenocytes in mediating lipid synthesis is more pronounced in newly eclosed adults when the adipose tissue needs to actively build up its lipid stores. In later stages of life, the lipid synthetic role of the oenocytes is repressed by the muscle-to-oenocyte Pvf1 signaling axis. This observation also raises the question of whether FFAs made in the oenocytes can be transported to the adipose tissue for storage. This possibility was tested by over-expressing the lipogenic genes fasn1 and fasn3, which regulate the rate limiting steps of de-novo lipid synthesis, in the oenocytes. It was found that excess lipids made in the oenocytes do end up in the adipose tissue of the animal leading to increased lipid droplet size in the adipose tissue. Taken together, these results provide evidence for the role of Drosophila oenocytes in lipid synthesis and storage of neutral lipids in the adipose tissue of the animal. Interestingly, the vertebrate liver is also one of the primary sites for de-novo lipid synthesis and lipids synthesized in the liver can be transported to the adipose tissue for the purpose of storage. Hence, the fundamental role of the oenocytes and the mammalian liver converge with respect to their involvement in lipid synthesis (Ghosh, 2020).
Oenocyte-specific loss of the components of the Pi3K/Akt1/TOR signaling pathway was observed to lead to increased lipid synthesis. The increased rate of lipid synthesis in flies lacking TOR signaling in the oenocytes is paradoxical to current knowledge of how TOR signaling affects expression of lipid synthesis genes. In both vertebrates and flies, TOR signaling is known to facilitate lipid synthesis by inducing the expression of key lipid synthesis genes such as acetyl CoA-carboxylase and fatty acid synthase via activation of SREBP-1 proteins. Therefore this study checked how oenocyte-specific loss of TOR signaling affects expression of oenocyte-specific fatty acid synthases (fasn2 and fasn3) and oenocyte non-specific fatty acid synthesis genes (fasn1 and acc). Oenocyte-specific loss of TOR strongly down-regulated only fasn2 and fasn3, while the expression of adipose tissue specific fasn1 and acc did not change, indicating that TOR signaling is required for the expression of lipogenic genes in the oenocytes. An increase in lipid synthesis in response to loss of TOR in the oenocytes is quite intriguing and the mechanism remains to be addressed. The increase in lipid synthesis is thought not to happen in the oenocytes since loss of TOR signaling rather reduces expression of lipogenic genes in the oenocytes. The increase in lipid synthesis could happen either as a result of compensatory upregulation of lipid synthesis in the adipose tissue or due to disruption of an as yet unknown role of the oenocytes in lipid synthesis that hinges on TOR signaling. The fact that the expression levels of fasn1 and acc does not change significantly in animals lacking TOR signaling in the oenocytes indicates that compensatory upregulation of lipid synthesis, if present, does not happen through transcriptional upregulation of lipid synthesis genes in the adipose tissue. It is still possible, however, that the increase in lipid synthesis is caused by post-translational modifications of the enzymes. Alternatively, loss of TOR in the oenocyte could affect tissue distribution of lipids or impair clearing of dietary lipids via formation of cuticular hydrocarbons. Understanding the tissue specific alterations in gene expression and changes in the phosphorylation states of key lipogenic proteins in the adipose tissue of animals lacking TOR signaling in oenocytes could shed more light on the mechanisms involved (Ghosh, 2020).
Interestingly, the data suggests that the Drosophila InR does not play a role in activating TOR signaling in the oenocytes. While loss of TOR signaling in the oenocytes leads to obesity, loss of InR signaling does not. Additionally, loss of oenocyte specific InR signaling did not have any effect on p4EBP levels in oenocytes. Moreover, InR signaling and TOR signaling also diverge in their roles in regulating the size of oenocytes. While loss of InR signaling leads to a significant reduction in the size of oenocytes, loss of TOR does not. Further suggesting that TOR does not act downstream of InR in oenocytes. Rather, the data suggests that in wildtype well-fed flies TOR signaling in oenocytes is activated by the Pvf receptor. Interestingly, insulin dependent activation of TOR is not universal. For instance, in the specialized cells of non-obese mouse liver, InR does not play any role in activation of TOR and downstream activation of SREBP-1c (Ghosh, 2020).
Drosophila larval oenocytes are known to accumulate lipids in response to starvation. It has also been showed that starving adult females for 36 hr is capable of inducing lipid accumulation in the oenocytes and that this response is dependent of InR signaling. Since TOR signaling is a known metabolic regulator, one alternate hypothesis that could explain some of the data is that loss of PvR/TOR signaling leads to a starvation like response specifically in the oenocyte leading to InR-dependent accumulation of lipid droplets. To address this possibility, single nuclei sequencing of the adult male abdominal cuticle (and the tissues residing within) derived from oenots>tsc1,tsc2 flies. The animals were raised under identical experimental conditions as control animals. Then the two snRNA-seq data sets were re-analyzed after correcting for batch effects using harmony. The resulting UMAP plots for both genotypes look similar to the original UMAP plot for the control flies and identifies all the clusters reported (see Differential snRNA-seq of the abdominal cuticle upon oenocyte-specific loss of TOR). The percentage of nuclei that constitute each of the major clusters remained similar in both genotypes and the top marker genes for each of the clusters did not change. The oenocyte-specific gene expression profiles from both data sets were subsequently converted to pseudobulk expression for the genes that were detected. This allowed comparison of the expression profiles of the oenocytes from control animals and animals lacking TOR signaling in oenocytes. The effect of losing TOR on the expression of the 47 genes that had been reported to be up-regulated in oenocytes in response to starvation was specifically looked at. Thirty-six of these genes were detected by single nuclei sequencing analysis, however, none of them changed significantly. Based on this observation, it is concluded that loss of TOR signaling most likely does not mount a starvation like response in the oenocytes (Ghosh, 2020).
Serum levels of VEGF-A is high in obese individuals and drops rapidly in response to bariatric surgery, suggesting a role for VEGF-A in obesity. However, evidence on whether VEGF-A or other VEGFs are deleterious vs beneficial in the context of the pathophysiology of obesity is unclear. Adipose tissue-specific over-expression of both VEGF-B and VEGF-A has been shown to improve adipose tissue vascularization, reduce hypoxia, induce browning of fat, increase thermogenesis, and protect against obesity. At the same time, blocking VEGF-A signaling in the adipose tissue of genetically obese mice leads to reduction of body weight gain, improvement in insulin sensitivity, and a decrease in adipose tissue inflammation. Moreover, systemic inhibition of VEGF-A or VEGF-B signaling by injecting neutralizing monoclonal antibodies have also shown remarkable effects in improving insulin sensitivity in the muscle, adipose tissue, and the liver of high-fat diet-induced mouse models of obesity and diabetes. Although the evidence on the roles of VEGF/PDGF signaling ligands in obesity and insulin resistance is well established, the mechanisms clearly are quite complex and are often context dependent. Consequently, a wider look at various tissue specific roles of PDGF/VEGF signaling will be necessary to comprehensively understand the roles of PDGF/VEGF signaling in lipid metabolism. The current work demonstrates an evolutionarily conserved role for PDGF/VEGF signaling in lipid metabolism and a non-endothelial cell dependent role of the signaling pathway in lipid synthesis. Additionally, these findings suggest an atypical tissue-specific role of TOR signaling in suppressing lipid synthesis at the level of the whole organism. Further studies will be required to determine whether vertebrate VEGF/PDGF and TOR signaling exerts similar roles either in the vertebrate liver or in other specialized organ (Ghosh, 2020).
This study made use of snRNA-Seq technology to identify expression of Pvr precisely in certain tissues in the complex abdominal region, which harbors several metabolically active tissues including adipose tissues, oenocytes, and muscle in Drosophila. As yet, there is no systematic study of a complete transcriptomics resource of each of these tissues considering the difficulty in dissecting and segregating these tissues for downstream sequencing. Thus, this study also provides a rich resource of gene expression profiles, paving way for a systems-level understanding of each of these tissues in Drosophila (Ghosh, 2020).
Correct orchestration of nervous system development is a profound challenge that involves coordination of complex molecular and cellular processes. Mechanistic target of rapamycin (mTOR) signaling is a key regulator of nervous system development and synaptic function. The mTOR kinase is a hub for sensing inputs including growth factor signaling, nutrients and energy levels. Activation of mTOR signaling causes diseases with severe neurological manifestations, such as tuberous sclerosis complex and focal cortical dysplasia. However, the molecular mechanisms by which mTOR signaling regulates nervous system development and function are poorly understood. Unkempt is a conserved zinc finger/RING domain protein that regulates neurogenesis downstream of mTOR signaling in Drosophila. Unkempt also directly interacts with the mTOR complex I component Raptor. This study described the generation and characterisation of mice with a conditional knockout of Unkempt (Unk(cKO)) in the nervous system. Loss of Unkempt reduces Raptor protein levels in the embryonic nervous system but does not affect downstream mTORC1 targets. It was also shown that nervous system development occurs normally in Unk(cKO) mice. However, it was found that Unkempt is expressed in the adult cerebellum and hippocampus and behavioural analyses show that Unk(cKO) mice have improved memory formation and cognitive flexibility to re-learn. Further understanding of the role of Unkempt in the nervous system will provide novel mechanistic insight into the role of mTOR signaling in learning and memory (Vinsland, 2021).
The gut is the primary interface between an animal and food, but how it adapts to qualitative dietary variation is poorly defined. This study finds that the Drosophila midgut plastically resizes following changes in dietary composition. A panel of nutrients collectively promote gut growth, which sugar opposes. Diet influences absolute and relative levels of enterocyte loss and stem cell proliferation, which together determine cell numbers. Diet also influences enterocyte size. A high sugar diet inhibits translation and uncouples intestinal stem cell proliferation from expression of niche-derived signals, but, surprisingly, rescuing these effects genetically was not sufficient to modify diet's impact on midgut size. However, when stem cell proliferation was deficient, diet's impact on enterocyte size was enhanced, and reducing enterocyte-autonomous TOR signaling was sufficient to attenuate diet-dependent midgut resizing. These data clarify the complex relationships between nutrition, epithelial dynamics, and cell size, and reveal a new mode of plastic, diet-dependent organ resizing (Bonfini, 2021).
Chronic exercise is widely recognized as an important contributor to healthspan in humans and in diverse animal models. Recently, it has been demonstrated that Sestrins, a family of evolutionarily conserved exercise-inducible proteins, are critical mediators of exercise benefits in flies and mice. Knockout of Sestrins prevents exercise adaptations to endurance and flight in Drosophila, and similarly prevents benefits to endurance and metabolism in exercising mice. In contrast, overexpression of dSestrin in muscle mimics several of the molecular and physiological adaptations characteristic of endurance exercise. This study extended those observations to examine the impact of dSestrin on preserving speed and increasing lysosomal activity. dSestrin was found to be a critical factor driving exercise adaptations to climbing speed, but is not absolutely required for exercise to increase lysosomal activity in Drosophila. The role of Sestrin in increasing speed during chronic exercise requires both the TORC2/AKT axis and the PGC1α homolog Spargel, while dSestrin requires interactions with TORC1 to cell-autonomously increase lysosomal activity. These results highlight the conserved role of Sestrins as key factors that drive diverse physiological adaptations conferred by chronic exercise (Sujkowski, 2021).
Most cells in a developing organ stop proliferating when the organ reaches a correct, final size. The underlying molecular mechanisms are not understood. This study finds that in Drosophila the hormone ecdysone controls wing disc size. To study how ecdysone affects wing size, endogenous ecdysone synthesis was inhibited and larvae were fed exogenous ecdysone in a dose-controlled manner. For any given ecdysone dose, discs stop proliferating at a particular size, with higher doses enabling discs to reach larger sizes. Termination of proliferation coincides with a drop in TORC1, but not Dpp or Yki signaling. Reactivating TORC1 bypasses the termination of proliferation, indicating that TORC1 is a main downstream effector causing proliferation termination at the maximal ecdysone-dependent size. Experimental manipulation of Dpp or Yki signaling can bypass proliferation termination in hinge and notum regions, but not the pouch, suggesting that the mechanisms regulating proliferation termination may be distinct in different disc regions (Strassburger, 2021).
When infected by enteric pathogenic bacteria, animals need to initiate local and whole-body defence strategies. While most attention has focused on the role innate immune anti-bacterial responses, less is known about how changes in host metabolism contribute to host defence. Using Drosophila as a model system, this study identified induction of intestinal target-of-rapamycin (TOR) kinase signaling as a key adaptive metabolic response to enteric infection. Enteric infection induces both local and systemic induction of TOR independently of the IMD innate immune pathway, and TOR functions together with IMD signaling to promote infection survival. These protective effects of TOR signaling are associated with re-modelling of host lipid metabolism. Thus, TOR is required to limit excessive infection-mediated wasting of host lipid stores by promoting an increase in the levels of gut- and fat body-expressed lipid synthesis genes. These data supports a model in which induction of TOR represents a host tolerance response to counteract infection-mediated lipid wasting in order to promote survival (Deshpande, 2022).
Animals must sense and respond to nutrient availability in their local niche. This task is coordinated in part by the mTOR complex 1 (mTORC1) pathway, which regulates growth and metabolism in response to nutrients. In mammals, mTORC1 senses specific amino acids through specialized sensors that act through the upstream GATOR1/2 signaling hub. To reconcile the conserved architecture of the mTORC1 pathway with the diversity of environments that animals can occupy, this study hypothesized that the pathway might maintain plasticity by evolving distinct nutrient sensors in different metazoan phyla. This study identified the Drosophila protein Unmet expectations (Unmet, as a species-restricted nutrient sensor and traced its incorporation into the mTORC1 pathway. Upon methionine starvation, Unmet binds to the fly GATOR2 complex to inhibit dTORC1. S -adenosylmethionine (SAM), a proxy for methionine availability, directly relieves this inhibition. Unmet expression is elevated in the ovary, a methionine-sensitive niche, and flies lacking Unmet fail to maintain the integrity of the female germline under methionine restriction. By monitoring the evolutionary history of the Unmet-GATOR2 interaction, this study shows that the GATOR2 complex evolved rapidly in Dipterans to recruit and repurpose an independent methyltransferase as a SAM sensor. Thus, the modular architecture of the mTORC1 pathway allows it to co-opt preexisting enzymes and expand its nutrient sensing capabilities, revealing a mechanism for conferring evolvability on an otherwise highly conserved system (Liu, 2023).
Target of Rapamycin Complex I (TORC1) is a central regulator of metabolism in eukaryotes that responds to a wide array of negative and positive inputs. The GTPase-activating protein toward Rags (GATOR) signaling pathway acts upstream of TORC1 and is comprised of two subcomplexes. The trimeric GATOR1 complex inhibits TORC1 activity in response to amino acid limitation by serving as a GTPase-activating protein (GAP) for the TORC1 activator RagA/B, a component of the lysosomally located Rag GTPase. The multi-protein GATOR2 complex inhibits the activity of GATOR1 and thus promotes TORC1 activation. This study reports that Wdr59, originally assigned to the GATOR2 complex based on studies performed in tissue culture cells, unexpectedly has a dual function in TORC1 regulation in Drosophila. ovary and the eye imaginal disc brain complex, Wdr59 inhibits TORC1 activity by opposing the GATOR2-dependent inhibition of GATOR1. Conversely, in the Drosophila fat body, Wdr59 promotes the accumulation of the GATOR2 component Mio and is required for TORC1 activation. Similarly, in mammalian HeLa cells, Wdr59 prevents the proteolytic destruction of GATOR2 proteins Mio and Wdr24. Consistent with the reduced levels of the TORC1-activating GATOR2 complex, Wdr59KOs HeLa cells have reduced TORC1 activity which is restored along with GATOR2 protein levels upon proteasome inhibition. Taken together, these data support the model that the Wdr59 component of the GATOR2 complex functions to promote or inhibit TORC1 activity depending on cellular context (Zhang, 2023).
The highly conserved Target of Rapamycin Complex 1 (TORC1) is a central regulator of growth and metabolism in eukaryotes. In the presence of positive upstream inputs, TORC1 promotes anabolic metabolism and growth by phosphorylating numerous downstream targets including S6K and 4EBP. Conversely, when exposed to negative cues, such as limited nutrients, or the lack of growth factors, cells downregulate TORC1 to activate catabolic metabolism and inhibit growth. The deregulation of TORC1 is implicated in a wide array of diseases including cancer, epilepsy, and aging. Thus, there is intense interest in obtaining a mechanistic understanding of how upstream signaling pathways regulate TORC1 activity (Zhang, 2023).
The Rag GTPase is a heterodimer comprised of a Rag A/B subunit and a RagC/D subunit, that recruits TORC1 to lysosomes for activation by the small GTPase Rheb, when the RagA/B component of the GTPase is in the GTP-bound state. The GTPase-activating protein toward Rags (GATOR) complex is an important upstream regulator of TORC1 that responds to the presence of nutrients. The GATOR complex, originally identified in yeast and named the Seh1 Associated (SEA) complex, is comprised of two subcomplexes, GATOR1 and GATOR2. The GATOR1 complex, which contains the proteins Nprl2, Nprl3, and DEPDC5/Iml1, inhibits TORC1 activity by serving as a GAP (GTPase-activating protein) for the lysosomally located Rag GTPase (Zhang, 2023).
The GATOR2 complex inhibits GATOR1 and thus serves to activate TORC1. However, the mechanism by which GATOR2 inhibits GATOR1 remains unknown. In its initial functional characterization in mammalian and Drosophila cultured cells, the GATOR2 complex was reported to contain five protein Mios/Mio, Seh1, Sec13, Wdr24, and Wdr59. In these studies, knockdowns of GATOR2 components resulted in the constitutive activation of GATOR1 and decreased TORC1 activity. Similarly, Drosophila mutants of the GATOR2 components Mio, Seh1, and Wdr24 exhibit decreased TORC1 activity and growth in the female germline. However, a recent study from Schizosaccharomyces pombe reported that SEA3/WDR59 inhibits TORC1 activity as a component of the GATOR1 complex. Notably, this is oppositive to the role assigned to Wdr59 based on studies in both human and Drosophila cultured cells. Additionally, deletions of Wdr59 in HEK293 cells and mouse embryonic fibroblasts result in a partial resistance to nutrient deprivation. Thus, the exact function of Wdr59 within the GATOR-TORC1 signaling pathway remains unclear (Zhang, 2023).
This study defines the in vivo requirement for the GATOR component Wdr59 in Drosophila. Wdr59 displays two distinct functions depending on cell type. First, in the ovary and the eye imaginal disc brain complex, Wdr59 acts upstream of the GATOR2 complex to inhibit TORC1 activity. Second, in the adult fat body and mammalian HeLa cells, Wdr59 protects the GATOR2 complex from proteolysis and promotes TORC1 activity. These data provide mechanistic insight into the complex role of the GATOR component Wdr59 in the tissue-specific regulation of TORC1 activity (Zhang, 2023).
The GATOR complex is an essential upstream regulator of TORC1 that is frequently mutated in human disease. This study describes a dual function for the GATOR component Wdr59 in TORC1 regulation in Drosophila. Surprisingly, Wdr59 functions were found to inhibit or promote TORC1 activity depending on cellular context. These studies broaden understanding of the GATOR-TORC1 signaling axis in metazoans and demonstrate the importance of examining metabolic regulation in vivo (Zhang, 2023).
In vivo studies often reveal tissue-specific and/or metabolic requirements for genes that are not observed in cell culture. In cultured cells from both mammals and Drosophila, RNAi depletions of wdr59 result in decreased TORC1 activity and slow growth, thus phenocopying depletions/knockouts of the GATOR2 components wdr24, seh1, and mio. Based on these initial studies, Wdr59 was assigned to the GATOR2 complex, which promotes TORC1 activity by downregulating the TORC1 inhibitor GATOR1. However, in vivo studies indicate wdr59 is not obligate member of the GATOR2 complex. Using Drosophila as a model, this study found that wdr59 mutant ovaries have increased TORC1 activity and growth rates and an attenuated response to nutrient stress. Notably, these phenotypes are the opposite of those reported for mutants of the GATOR2 components mio, seh1, and wdr24, but phenocopy those observed in mutants of the GATOR1 components nprl2, nprl3, and iml1. Thus, in the Drosophila ovary, Wdr59 functions to restrict TORC1 activity. In line with these findings, the binding of RagAGTP to the Nprl2 component of the GATOR1 complex was demonstrated to be decreased in wdr59 mutants, again the opposite of what was observed in mutants of the GATOR2 component wdr24. Taken together, these data suggest that wdr59 inhibits TORC1 activity by increasing the activity of the TORC1 inhibitor GATOR1. Notably, recent evidence from s. pombe proposes that Wdr59, known as SEA3 in yeast, acts to inhibit TORC1 as a component of the GATOR1 complex. However, epistasis analysis indicated that in Drosophila, Wdr59 is not a component of the GATOR1 complex but instead functions as an inhibitor of GATOR2 (Zhang, 2023).
How might wdr59 impact TORC1 activity? Biochemical, structural, and computational analysis from yeast indicate that SEA3/wdr59 is well positioned to function as an interacting hub connecting GATOR2 with GATOR1. Consistent with this observation, this study found that, as is observed in fission yeast, wdr59 regulates the interaction between components of the GATOR1 and GATOR2 complexes, with an increased association of GATOR2 with GATOR1 observed in the wdr59 mutant background. Thus, one possible model to explain these data is that wdr59 inhibits TORC1 activity by restricting the interaction of the GATOR2 complex with GATOR1, thus unleashing its GAP activity and TORC1 inhibitory potential. A prediction from this model is that components of the GATOR2 complex will be epistatic to Wdr59. In other words, Wdr59 requires the presence of an active GATOR2 complex to regulate TORC1 activity. Consistent with this prediction, it was fond that in the Drosophila ovary, wdr59, GATOR2 double mutants have a GATOR2-like phenotypes with reduced growth and TORC1 activity, strongly suggesting that GATOR2 is downstream of Wdr59. In contrast, in mutants or depletions of the GATOR1 components nprl2, nprl3, and Iml1, RagA remains in its TORC1-activating GTP-bound state, promoting the constitutive recruitment and activation of TORC1 on lysosomes independent of the status of GATOR2. Thus, GATOR1 components, unlike Wdr59, are epistatic to upstream members of the pathway including components of the GATOR2 complex. Taken together these data support the model that in Drosophila, Wdr59 is not required for GATOR1 function but instead acts upstream of the complex to regulate the activity of the GATOR1 inhibitor GATOR2. Importantly, the data demonstrate that the GATOR2 complex can inhibit GATOR1 independent of the Wdr59 subunit (Zhang, 2023).
Currently, there are conflicting reports on the role of Wdr59/Sea3 in the regulation of TORC1 activity. In Drosophila and mammalian cultured cells, as well as in mouse breast cancer tumors, Wdr59 promotes TORC1 activity while in fission yeast Wdr59 inhibits TORC1 activity. The current results provide a potential explanation for this contradiction. In Drosophila, Wdr59 either promotes or inhibits TORC1 activity depending on cell type. In the female germline and the eye imaginal disc brain complex, Wdr59 inhibits TORC1 activity. However, in the adult fat body, Wdr59 promotes TORC1 activity. Importantly, in the adult fat body, but not the ovary, Wdr59 is required for the accumulation of the Mio protein. Thus, a possible reason that wdr59 mutant fat bodies have reduced TORC1 activity is that they do not have a functional GATOR2 complex. This model is consistent with the finding that the GATOR2 complex is downstream of Wdr59 (Zhang, 2023).
To further explore why Wdr59 promotes TORC1 activation in some cellular contexts, the levels of GATOR2 proteins were examined in Wdr59KO HeLa cells. It was wondered if the TORC1 promoting function for Wdr59 observed in cultured cells might reflect the requirement for Wdr59 protein to accumulate components of the GATOR2 complex. Indeed, dramatically lower levels of the GATOR2 components Mios and Wdr24 were found in Wdr59KO HeLa cells due to proteolytic destruction by the proteosome. Strikingly, inhibiting the proteasome pharmacologically resulted in increased levels of GATOR2 proteins which was accompanied by a partial rescue of TORC1 activity. These data strongly suggest that the low TORC1 activity observed in Wdr59KD or Wdr59KO HeLa cells is due at least in part to the concomitant decrease in GATOR2 protein levels (Zhang, 2023).
Taken together, these data support the model that Wdr59 either promotes or inhibits TORC1 activity depending on cellular context. The GATOR-TORC1 signaling pathway is frequently cited as a potential target of pharmaceutical intervention because of its role in cancer and epilepsy. Thus, it is essential to have a full mechanistic understanding of the in vivo function of the GATOR complex in the regulation of TORC1 signaling and growth in metazoans (Zhang, 2023).
Prior to submission of this manuscript, a detailed cryo-electron microscopy structure of the human GATOR2 complex was published. It was reported that GATOR2 is a large 1.1 Mda complex that forms a cage-like structure, built on a continuous scaffold. As previously described, GATOR2 complex components contain numerous features common to membrane coating complexes which can form scaffolds that alter the curvature of membranes. Consistent with these studies, the authors report that the two copies of the Wdr59 subunit mediate the association of GATOR2 with GATOR1. However, as discussed above, it was found that Wdr59 opposes the association of GATOR2 with GATOR1 in several Drosophila tissues, the opposite of what is reported in this study in HEK293T cells. Whole animal studies in Drosophila have determined that there are unique tissue-specific requirements for multiple individual GATOR2 subunits, including Mio, Seh1, Wdr24, and now Wdr59. Going forward, it will be fascinating to determine how GATOR2 structure mediates tissue-specific GATOR2 functions in vivo (Zhang, 2023).
Growth deceleration before growth termination is a universal feature of growth during development. Transcriptomics analysis reveals that during their two-day period of growth deceleration, wing imaginal discs of Drosophila undergo a progressive metabolic shift from oxidative phosphorylation towards glycolysis. Ultra-sensitive reporters of HIF-1alpha stability and activity show that imaginal discs become increasingly hypoxic during development in normoxic conditions, suggesting that limiting oxygen supply could underlie growth deceleration. This study confirm the expectation that rising levels of HIF-1alpha dampen TOR signalling activity through transcriptional activation of REDD1 (Scylla and Charybdis) . Conversely, excess TOR leads, in a tissue-size-dependent manner, to hypoxia, which boosts HIF-1alpha levels and activity. Thus, HIF-1alpha mediates a negative feedback loop whereby TOR signalling triggers hypoxia, which in turn reduces TOR signalling. Abrogation of this feedback by Sima/HIF-1alpha knockdown leads to cellular stress, which is alleviated by reduced TOR signalling or a modest increase in environmental oxygen. It is concluded that Sima/HIF-1alpha prevents TOR-mediated growth from depleting local oxygen supplies during normal development (Zhzo, 2025).
Nutrition shapes a broad range of life-history traits, ultimately impacting animal fitness. A key fitness-related trait, female fecundity is well known to change as a function of diet. In particular, the availability of dietary protein is one of the main drivers of egg production, and in the absence of essential amino acids egg laying declines. However, it is unclear whether all essential amino acids have the same impact on phenotypes like fecundity. Using a holidic diet, this study fed adult female Drosophila melanogaster diets that contained all necessary nutrients except one of the 10 essential amino acids and assessed the effects on egg production. For most essential amino acids, depleting a single amino acid induced as rapid a decline in egg production as when there were no amino acids in the diet. However, when either methionine or histidine were excluded from the diet, egg production declined more slowly. Next, this study tested whether GCN2 and TOR mediated this difference in response across amino acids. While mutations in GCN2 did not eliminate the differences in the rates of decline in egg laying among amino acid drop-out diets, it was found that inhibiting TOR signalling caused egg laying to decline rapidly for all drop-out diets. TOR signalling does this by regulating the yolk-forming stages of egg chamber development. These results suggest that amino acids differ in their ability to induce signalling via the TOR pathway. This is important because if phenotypes differ in sensitivity to individual amino acids, this generates the potential for mismatches between the output of a pathway and the animal's true nutritional status (Alves, 2022).
The mTORC1 nutrient-sensing pathway integrates metabolic and endocrine signals into the brain to evoke physiological responses to food deprivation, such as autophagy. Nevertheless, the impact of neuronal mTORC1 activity on neuronal circuits and organismal metabolism remains obscure. This study shows that mTORC1 inhibition acutely perturbs serotonergic neurotransmission via proteostatic alterations evoked by the autophagy inducer Atg1. Neuronal ATG1 alters the intracellular localization of the serotonin transporter, which increases the extracellular serotonin and stimulates the 5HTR7 postsynaptic receptor. 5HTR7 enhances food-searching behaviour and ecdysone-induced catabolism in Drosophila. Along similar lines, the pharmacological inhibition of mTORC1 in zebrafish also stimulates food-searching behaviour via serotonergic activity. These effects occur in parallel with neuronal autophagy induction, irrespective of the autophagic activity and the protein synthesis reduction. In addition, ectopic neuronal atg1 expression enhances catabolism via insulin pathway downregulation, impedes peptidergic secretion, and activates non-cell autonomous cAMP/PKA. The above exert diverse systemic effects on organismal metabolism, development, melanisation, and longevity. It is concluded that neuronal atg1 aligns neuronal autophagy induction with distinct physiological modulations, to orchestrate a coordinated physiological response against reduced mTORC1 activity.
Alves, A. N., Sgro, C. M., Piper, M. D. W. and Mirth, C. K. (2022). Target of Rapamycin Drives Unequal Responses to Essential Amino Acid Depletion for Egg Laying in Drosophila Melanogaster. Front Cell Dev Biol 10: 822685. PubMed ID: 35252188
Amoyel, M., Hillion, K. H., Margolis, S. R. and Bach, E. A. (2016). Somatic stem cell differentiation is regulated by PI3K/Tor signaling in response to local cues. Development 143(21):3914-3925 PubMed ID: 27633989
Bar-Peled, L., Schweitzer, L. D., Zoncu, R. and Sabatini, D. M. (2012). Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150(6): 1196-1208. PubMed ID: 22980980
Bar-Peled, L., Chantranupong, L., Cherniack, A. D., Chen, W. W., Ottina, K. A., Grabiner, B. C., Spear, E. D., Carter, S. L., Meyerson, M. and Sabatini, D. M. (2013). A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340(6136): 1100-1106. PubMed ID: 23723238
Bonfini, A., Dobson, A. J., Duneau, D., Revah, J., Liu, X., Houtz, P. and Buchon, N. (2021). Multiscale analysis reveals that diet-dependent midgut plasticity emerges from alterations in both stem cell niche coupling and enterocyte size. Elife 10. PubMed ID: 34553686
Cheng, Y., Cai, J., Fu, Y., Feng, C., Hao, Y. and Wei, Y. (2020). Royal jelly attenuates metabolic defects in a Drosophila mutant with elevated TORC1 activity. Biol Open 9(11). PubMed ID: 33037015
Demetriades, C., Doumpas, N. and Teleman, A. A. (2014). Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156: 786-799. PubMed ID: 24529380
Deshpande, R., Lee, B., Qiao, Y. and Grewal, S. S. (2022). TOR signaling is required for host lipid metabolic remodelling and survival following enteric infection in Drosophila. Dis Model Mech. PubMed ID: 35363274
Devilliers, M., Garrido, D., Poidevin, M., Rubin, T., Le Rouzic, A. and Montagne, J. (2021). Differential metabolic sensitivity of insulin-like-response- and TORC1-dependent overgrowth in Drosophila fat cells. Genetics 217(1): 1-12. PubMed ID: 33683355
Du, S., Zeng, S., Song, L., Ma, H., Chen, R., Luo, J., Wang, X., Ma, T., Xu, X., Sun, H., Yi, P., Guo, J., Huang, Y., Liu, M., Wang, T., Liao, W. P., Zhang, L., Liu, J. Y. and Tang, B. (2023). Functional characterization of novel NPRL3 mutations identified in three families with focal epilepsy. Sci China Life Sci. PubMed ID: 37071290
Frias, M. A., Mukhopadhyay, S., Lehman, E., Walasek, A., Utter, M., Menon, D. and Foster, D. A. (2019). Phosphatidic acid drives mTORC lysosomal translocation in the absence of amino acids. J Biol Chem 295(1):263-274. PubMed ID: 31767684
Ghosh, A. C., Tattikota, S. G., Liu, Y., Comjean, A., Hu, Y., Barrera, V., Ho Sui, S. J. and Perrimon, N. (2020). Drosophila PDGF/VEGF signaling from muscles to hepatocyte-like cells protects against obesity. Elife 9. PubMed ID: 33107824
Haller, S., Kapuria, S., Riley, R. R., O'Leary, M. N., Schreiber, K. H., Andersen, J. K., Melov, S., Que, J., Rando, T. A., Rock, J., Kennedy, B. K., Rodgers, J. T. and Jasper, H. (2017). mTORC1 activation during repeated regeneration impairs somatic stem cell maintenance. Cell Stem Cell 21(6): 806-818.e805. PubMed ID: 29220665
Han, J. M., Jeong, S. J., Park, M. C., Kim, G., Kwon, N. H., Kim, H. K., Ha, S. H., Ryu, S. H. and Kim, S. (2012). Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149(2): 410-424. PubMed ID: 22424946
Kim, A. R. and Choi, K. W. (2019). TRiC/CCT chaperonins are essential for organ growth by interacting with insulin/TOR signaling in Drosophila. Oncogene 38(24): 4739-4754. PubMed ID: 30792539
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P. and Guan, K. L. (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10: 935-945. PubMed ID: 18604198
Kim, M. J. and O'Connor, M. B. (2021). Drosophila Activin signaling promotes muscle growth through InR/TORC1-dependent and -independent processes. Development 148(1). PubMed ID: 33234715
Li, Y., Geng, J., Rimal, S., Wang, H., Liu, X., Lu, B. and Li, S. (2023). The mTORC2/AKT/VCP axis is associated with quality control of the stalled translation of poly(GR) dipeptide repeats in C9-ALS/FTD. J Biol Chem 299(3): 102995. PubMed ID: 36764521
Li, Z., Qian, W., Song, W., Zhao, T., Yang, Y., Wang, W., Wei, L., Zhao, D., Li, Y., Perrimon, N., Xia, Q. and Cheng, D. (2022). A salivary gland-secreted peptide regulates insect systemic growth. Cell Rep 38(8): 110397. PubMed ID: 35196492
Liu, G. Y., Jouandin, P., Bahng, R. E., Perrimon, N. and Sabatini, D. M. (2023). An evolutionary mechanism to assimilate new nutrient sensors into the mTORC1 pathway. bioRxiv. PubMed ID: 37292894
Liu, P., Guo, Y., Xu, W., Song, S., Li, X., Wang, X., Lu, J., Guo, X., Richardson, H. E. and Ma, X. (2022). Ptp61F integrates Hippo, TOR, and actomyosin pathways to control three-dimensional organ size. Cell Rep 41(7): 111640. PubMed ID: 36384105
Lottes, E. N., Ciger, F. H., Bhattacharjee, S., Timmins-Wilde, E. A., Tete, B., Tran, T., Matta, J., Patel, A. A. and Cox, D. N. (2023). CCT and Cullin1 regulate the TORC1 pathway to promote dendritic arborization in health and disease. bioRxiv. PubMed ID: 37577581
Metaxakis, A., Pavlidis, M., Tavernarakis, N. (2023). Neuronal atg1 Coordinates Autophagy Induction and Physiological Adaptations to Balance mTORC1 Signalling. Cells, 12(16) PubMed ID: 37626835
Nishida, H., Okada, M., Yang, L., Takano, T., Tabata, S., Soga, T., Ho, D. M., Chung, J., Minami, Y. and Yoo, S. K. (2021). Methionine restriction breaks obligatory coupling of cell proliferation and death by an oncogene Src in Drosophila. Elife 10. PubMed ID: 33902813
Ohhara, Y., Hoshino, G., Imahori, K., Matsuyuki, T. and Yamakawa-Kobayashi, K. (2021). The Nutrient-Responsive Molecular Chaperone Hsp90 Supports Growth and Development in Drosophila. Front Physiol 12: 690564. PubMed ID: 34239451
Ovrebo, J. I., Bradley-Gill, M. R., Zielke, N., Kim, M., Marchetti, M., Bohlen, J., Lewis, M., van Straaten, M., Moon, N. S. and Edgar, B. A. (2022). Translational control of E2f1 regulates the Drosophila cell cycle. Proc Natl Acad Sci U S A 119(4). PubMed ID: 35074910
Parkhitko, A. A., Dambowsky, M., Asara, J. M., Nemazanyy, I., Dibble, C. C., Simons, M. and Perrimon, N. (2022). Lysosomal cystine mobilization shapes the response of TORC1 and tissue growth to fasting. Science 375(6582): eabc4203. PubMed ID: 35175796
Perez-Gomez, R., Magnin, V., Mihajlovic, Z., Slaninova, V. and Krejci, A. (2020). Downregulation of respiratory complex I mediates major signalling changes triggered by TOR activation. Sci Rep 10(1): 4401. PubMed ID: 32157127
Petit, C. S., Roczniak-Ferguson, A. and Ferguson, S. M. (2013). Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J Cell Biol 202(7): 1107-1122. PubMed ID: 24081491
Rambur, A., Lours-Calet, C., Beaudoin, C., Bunay, J., Vialat, M., Mirouse, V., Trousson, A., Renaud, Y., Lobaccaro, J. A., Baron, S., Morel, L. and de Joussineau, C. (2020). Sequential Ras/MAPK and PI3K/AKT/mTOR pathways recruitment drives basal extrusion in the prostate-like gland of Drosophila. Nat Commun 11(1): 2300. PubMed ID: 32385236
Rebelo, A. R. and Homem, C. C. F. (2023). dMyc-dependent upregulation of CD98 amino acid transporters is required for Drosophila brain tumor growth. Cell Mol Life Sci 80(1): 30. PubMed ID: 36609617
Rodrigues, N. R., Macedo, G. E., Martins, I. K., Vieira, P. B., Kich, K. G., Posser, T., Franco, J. L. (2023). Sleep disturbance induces a modulation of clock gene expression and alters metabolism regulation in Drosophila. Physiology & behavior, 271:114334 PubMed ID: 37595818
Sancak, Y., Peterson, T. R., Shaul, Y. D., Lindquist, R. A., Thoreen, C. C., Bar-Peled, L. and Sabatini, D. M. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496-1501. PubMed ID: 18497260
Song, W., Kir, S., Hong, S., Hu, Y., Wang, X., Binari, R., Tang, H. W., Chung, V., Banks, A. S., Spiegelman, B. and Perrimon, N. (2019). Tumor-derived ligands trigger tumor growth and host wasting via differential MEK activation. Dev Cell 48(2): 277-286 e276. PubMed ID: 30639055
Storelli, G., Nam, H. J., Simcox, J., Villanueva, C. J. and Thummel, C. S. (2019). Drosophila HNF4 directs a switch in lipid metabolism that supports the transition to adulthood. Dev Cell 48(2): 200-214 e206. PubMed ID: 30554999
Strassburger, K., Lutz, M., Muller, S. and Teleman, A. A. (2021). Ecdysone regulates Drosophila wing disc size via a TORC1 dependent mechanism. Nat Commun 12(1): 6684. PubMed ID: 34795214
Strilbytska, O. M., Storey, K. B. and Lushchak, O. V. (2020). TOR signaling inhibition in intestinal stem and progenitor cells affects physiology and metabolism in Drosophila. Comp Biochem Physiol B Biochem Mol Biol 243-244: 110424. PubMed ID: 32088257
Szlachcic, E., Labecka, A. M., Privalova, V., Sikorska, A. and Czarnoleski, M. (2023). Systemic orchestration of cell size throughout the body: influence of sex and rapamycin exposure in Drosophila melanogaster. Biol Lett 19(3): 20220611. PubMed ID: 36946132
Sujkowski, A. and Wessells, R. (2021). Exercise and Sestrin Mediate Speed and Lysosomal Activity in Drosophila by Partially Overlapping Mechanisms.
Cells 10(9). PubMed ID: 34572128
Tandon, S. and Sarkar, S. (2021). The S6k/4E-BP mediated growth promoting sub-pathway of insulin signalling cascade is essential to restrict pathogenesis of poly(Q) disorders in Drosophila. Life Sci 275: 119358. PubMed ID: 33744321
Tang, H. W., Weng, J. H., Lee, W. X., Hu, Y., Gu, L., Cho, S., Lee, G., Binari, R., Li, C., Cheng, M. E., Kim, A. R., Xu, J., Shen, Z., Xu, C., Asara, J. M., Blenis, J. and Perrimon, N. (2021). mTORC1-chaperonin CCT signaling regulates m(6)A RNA methylation to suppress autophagy. Proc Natl Acad Sci U S A 118(10). PubMed ID: 33649236
Tang, X., Zhang, Y., Wang, G., Zhang, C., Wang, F., Shi, J., Zhang, T. and Ding, J. (2022). Molecular mechanism of S-adenosylmethionine sensing by SAMTOR in mTORC1 signaling. Sci Adv 8(26): eabn3868. PubMed ID: 35776786
Texada, M. J., Lassen, M., Pedersen, L. H., Koyama, T., Malita, A. and Rewitz, K. (2022). Insulin signaling couples growth and early maturation to cholesterol intake in Drosophila. Curr Biol 32(7): 1548-1562. PubMed ID: 35245460
Tiebe, M., Lutz, M., De La Garza, A., Buechling, T., Boutros, M. and Teleman, A. A. (2015). Reptor and Reptor-BP regulate organismal metabolism and transcription downstream of TORC1. Dev Cell 33: 272-284. PubMed ID: 25920570
Tsokanos, F. F., Albert, M. A., Demetriades, C., Spirohn, K., Boutros, M. and Teleman, A. A. (2016). eIF4A inactivates TORC1 in response to amino acid starvation. EMBO J 35(10):1058-76. PubMed ID: 26988032
Tsun, Z. Y., Bar-Peled, L., Chantranupong, L., Zoncu, R., Wang, T., Kim, C., Spooner, E. and Sabatini, D. M. (2013). The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 52(4): 495-505. PubMed ID: 24095279
Vinayagam, A., Kulkarni, M. M., Sopko, R., Sun, X., Hu, Y., Nand, A., Villalta, C., Moghimi, A., Yang, X., Mohr, S. E., Hong, P., Asara, J. M. and Perrimon, N. (2016). An integrative analysis of the InR/PI3K/Akt network identifies the dynamic response to insulin signaling. Cell Rep 16(11): 3062-3074. PubMed ID: 27626673
Vinsland, E., Baskaran, P., Mihaylov, S. R., Hobbs, C., Wood, H., Bouybayoune, I., Shah, K., Houart, C., Tee, A. R., Murn, J., Fernandes, C. and Bateman, J. M. (2021). The zinc finger/RING domain protein Unkempt regulates cognitive flexibility. Sci Rep 11(1): 16299. PubMed ID: 34381067
Xia, B., Viswanatha, R., Hu, Y., Mohr, S. E. and Perrimon, N. (2023). Pooled genome-wide CRISPR activation screening for rapamycin resistance genes in Drosophila cells. Elife 12. PubMed ID: 37078570
Yang, G., Humphrey, S. J., Murashige, D. S., Francis, D., Wang, Q. P., Cooke, K. C., Neely, G. and James, D. E. (2018). RagC phosphorylation autoregulates mTOR complex 1. EMBO J. PubMed ID: 30552228
Yang, S., Zhang, Y., Ting, C. Y., Bettedi, L., Kim, K., Ghaniam, E. and Lilly, M. A. (2020). The Rag GTPase regulates the dynamic behavior of TSC downstream of both amino acid and growth factor restriction. Dev Cell. PubMed ID: 32898476
Wei, Y., Bettedi, L., Ting, C. Y., Kim, K., Zhang, Y., Cai, J. and Lilly, M. A. (2019). The GATOR complex regulates an essential response to meiotic double-stranded breaks in Drosophila. Elife 8. PubMed ID: 31650955
Zhang, Y., Ting, C. Y., Yang, S., Reich, J., Fru, K. and Lilly, M. A. (2023). Wdr59 promotes or inhibits TORC1 activity depending on cellular context. Proc Natl Acad Sci U S A 120(1): e2212330120. PubMed ID: 36577058
Zhou, Y., Guan, J., Meng, G., Fan, W., Ge, C., Niu, C., Cheng, Y., Fu, Y., Lu, Y. and Wei, Y. (2023). The RagA GTPase protects young egg chambers in Drosophila. Cell Rep 42(6): 112631. PubMed ID: 37302067
Home page: The Interactive Fly © 1995, 1996 Thomas B. Brody, Ph.D.
The Interactive Fly resides on the
Society for Developmental Biology's Web server.
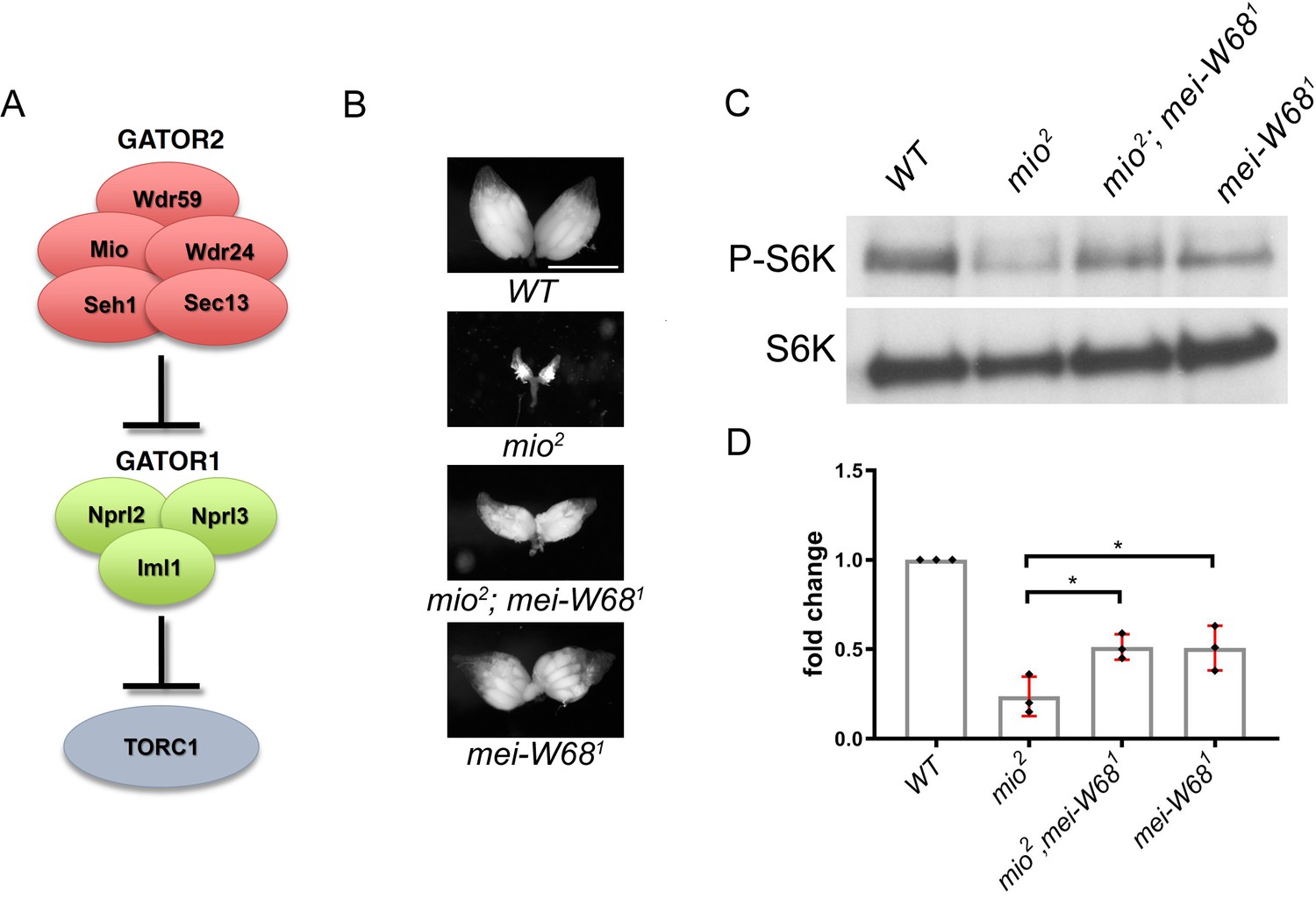{kind=link}
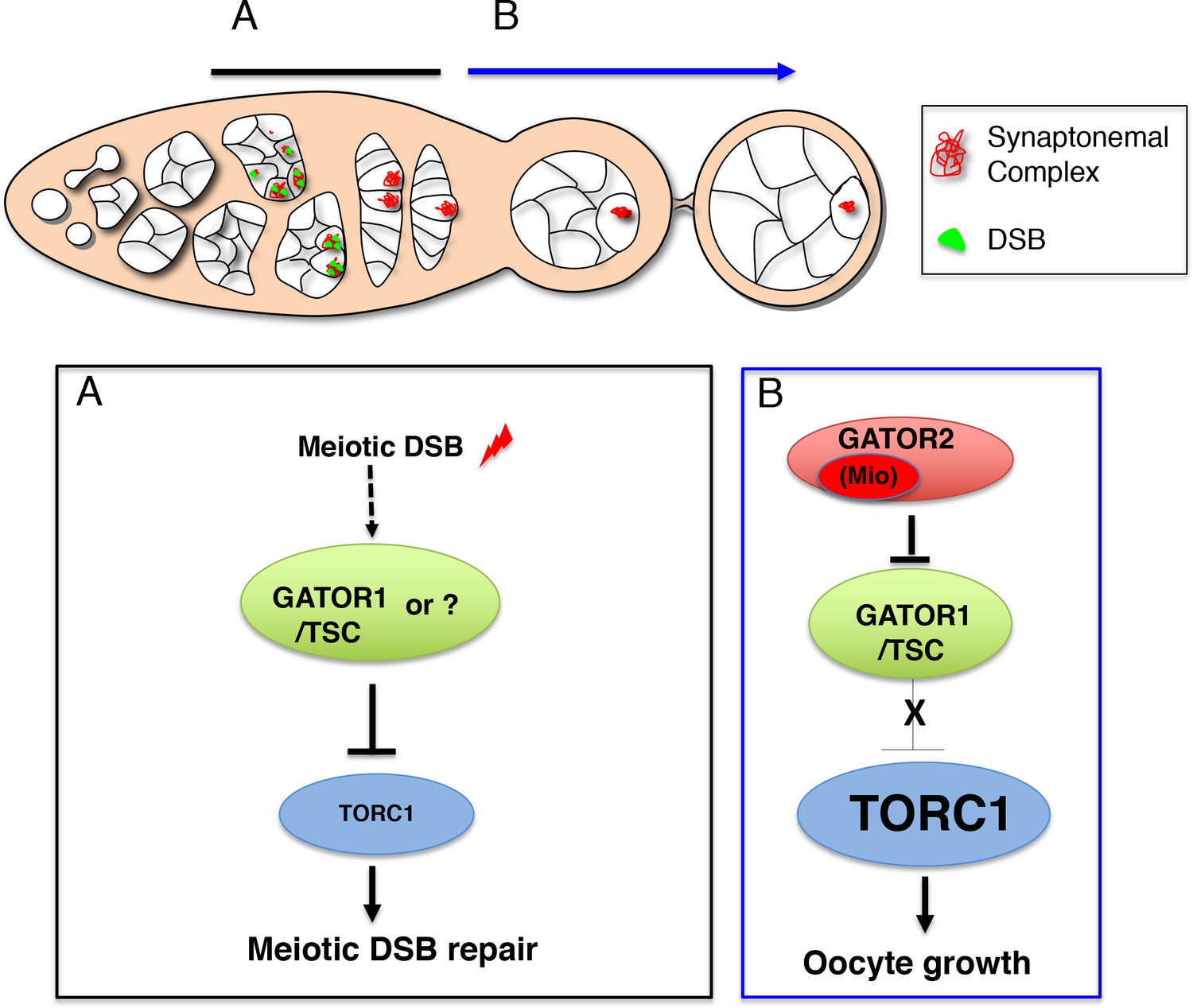{kind=link}
{kind=link}