The Interactive Fly
Zygotically transcribed genes
Sex Determination and Dosage Compensation Genes
|
The Interactive Fly Sex Determination and Dosage Compensation Genes |
|
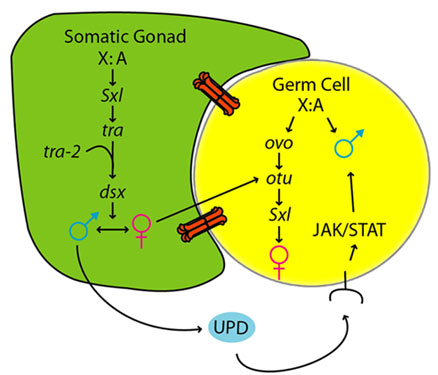
| A simplified model for sex determination in the somatic gonad and germline |
The variety of primary sex determination cues was appreciated long before the advent of molecular genetics. The two broadest categories are genetic sex determination (GSD), in which the sex of offspring is set by a sex chromosome or an autosomal gene, and environmental sex determination (ESD), in which sex is determined by temperature (as with turtles), local sex ratio (as with some tropical fish), or population density (as with mermithid nematodes). Though little is known about the molecular mechanisms of ESD, within the GSD systems many different mechanisms have been uncovered. Dual sex chromosome systems, in which either the female (ZW/ZZ) or the male (XX/XY) is heterogametic, are common, as are systems set by the ratio of the number of X chromosomes to sets of autosomes (X:A). There are also systems in which heterozygosity at a single locus is required for female development (known as complementary sex determination), as well as systems involving sex determination via multiple genes with additive effects (Haag, 2005; see full text of article).
Molecular genetic investigations of GSD in model systems such as Drosophila, Caenorhabditis, and mice have revealed a clear lack of conservation, underscoring the diversity. For example, although the primary sex determination signal in both D. melanogaster and C. elegans is the X:A ratio, the fruit fly pathway consists of a cell-autonomous cascade of regulated mRNA splicing, while that of the nematode follows a Hedgehog-like intercellular signaling pathway. GSD in mammals depends (with some interesting exceptions upon a Y-specific dominant gene (Sry) encoding a transcription factor. In the face of such impressive differences, perhaps the assumption of homology should be questioned: could it be that sex determination in different taxa has arisen independently over and over again in evolution? Until 1998, this seemed like a good bet (Haag, 2005).
The discovery of the homology of the key sex-determining genes doublesex in Drosophila and mab-3 in C. elegans provided the first evidence for a common evolutionary basis of sex determination in animals. Soon, related doublesex-mab-3 (DM)-family genes with roles in male sexual development were discovered in vertebrates and even cnidarians. Here at last was a smoking gun that could link the diverse metazoan sex determination systems. But as satisfying as the result was, it immediately gave birth to another mystery: if the enormous diversity of sex determination systems are all derived from a common ancestor, how could they possibly have been modified so radically? After all, sexual differentiation and reproduction are hardly unimportant developmental processes (Haag, 2005).
To understand how such diversity came to be, differences between closely related species must be examined. This approach allows the discovery and interpretation of small-scale sex determination changes before they are obscured by subsequent changes. The processes discovered in this way might then be reasonably extrapolated to explain the seemingly unrelated systems of more deeply diverged taxa. Work in dipterans has revealed three evolutionary phenomena that characterize shorter-term sex determination evolution (Haag, 2005).
The first of these is the often astounding rate of molecular evolution at the level of nucleotide and aminoacid sequences. Although some sex-determining genes are well conserved, many show unprecedented substitution rates. An extreme example is the central integrator of the X:A ratio in Caenorhabditis, xol-1. The xol-1 orthologues of the closely related nematodes C. elegans and C. briggsae are a mere 22% identical, even though genes surrounding xol-1 are much better conserved. Remarkably, the 3′ neighbor of xol-1, the immunoglobulin dim-1, is only 5 kb away and is essentially identical between species (Haag, 2005).
A second phenomenon, best exemplified by dipteran insects, is the modification of genetic control pathways through the gain or loss of key pathway components. In Drosophila, the first gene to respond to the X:A ratio is Sxl, whose transcription is regulated by both autosomal and X-linked factors very early in development. When X: A = 1 (i.e., in female embryos), Sxl transcription occurs and produces Sxl protein. Later in development, transcription from a second promoter occurs in both sexes, but these transcripts cannot be productively spliced without the earlier burst of Sxl expression. As a result, only females sustain Sxl expression, and in turn only females can productively splice the mRNA of tra, its downstream target. Productive splicing of tra is required to produce the female-specific form of dsx, a founding member of the DM family mentioned above (Haag, 2005).
In a series of groundbreaking papers, Saccone and colleagues investigated the pathway in the more distantly related heterogametic Mediterranean fruit fly Ceratitis capitata. The first surprise was that although a highly conserved Sxl homologue exists in Ceratitis, it does not undergo sex-specific regulation similar to that of Drosophila, which suggests that it does not play a key switch role (Saccone, 1998). Similar results have also been found for the housefly, Musca domestica, indicating that the role of Sxl in sex determination may be restricted to Drosophila and its closest relatives. In contrast, tra and dsx are key sex regulators in all dipterans examined thus far (Haag, 2005).
A further surprise came when the Ceratitis tra homologue was characterized. In the case of this gene, clear evidence for sex-specific regulation was found, and as with Drosophila, only females productively splice tra mRNA. However, this splicing difference can be explained nicely by a positive feedback, similar to that seen in Drosophila Sxl, in which Tra protein regulates its own splicing. It has been proposed that the dominant, male-specifying M factor on the Y chromosome inhibits this autoregulation. As a result, males cannot make functional Tra protein, and the male form of Dsx is produced. These experiments show not only how a pathway can evolve, but also, importantly, how X:A and heterogametic GSD systems can be interconverted by modifying the cue that regulates a conserved molecular switch gene (the splicing of tra mRNA) (Haag, 2005).
Finally, recent studies of Caenorhabditis nematodes have shed light on the genetic basis of the convergent evolution of sex determination related to mating system adaptations. An important factor in this area are new phylogenies of the genus, which consistently suggest the surprising possibility that the closely related hermaphroditic species C. elegans and C. briggsae acquired self-fertilization independently, from distinct gonochoristic (male/female) ancestors. Although this scenario is somewhat uncertain purely on parsimony grounds, recent work on the genetic control of the germline bisexuality that defines hermaphroditism has tipped the balance toward parallel evolution (Haag, 2005).
C. elegans fog-2, a gene required for spermatogenesis in hermaphrodites but not in males, has been cloned. It became clear that fog-2 is part of a large family of F-box genes and was produced by several recent rounds of gene duplication. The C. briggsae genome sequence suggested that while C. briggsae possesses a similarly large family of F-box proteins, the duplication event giving rise to fog-2 was specific to the C. elegans lineage. This work has been extended by the rigorous demonstration that fog-2 is indeed absent in C. briggsae. A short, C-terminal domain has been identified that makes FOG-2 uniquely able to perform its germline sex-determining function. This domain is probably derived from a frame-shifting mutation in an ancestral gene. Working with C. briggsae, evidence has been found of important species-specific regulation of germline sex determination. RNA interference and gene knockout approaches have shown that while C. elegans requires the male-promoting genes fem-2 and fem-3 to produce sperm in hermaphrodites, C. briggsae requires neither. Given that both genes have conserved roles in male somatic sex determination, this suggests that C. briggsae evolved hermaphroditism in a way that bypasses these genes (Haag, 2005).
The long-standing mystery of sex determination and its diversity began by comparisons between distantly related species. Recent work on closer relatives has uncovered processes that through a reasonable extrapolation enable the connection of these disparate dots into a fascinating picture of developmental evolution. Though the divergence is extreme, it is likely that a better understanding of the evolution of sex determination genes and pathways holds lessons about the evolution of development in general. The next major challenge will be to integrate the comparative developmental data with the ecological and population processes that are driving the evolution of sex determination. Only then will it be possible to say that the picture is complete (Haag, 2005).
The X chromosome of Drosophila shows a deficiency of genes with male-biased expression (see Sturgill, 2007), whereas mammalian X chromosomes are enriched for spermatogenesis genes expressed premeiosis and multicopy testis genes. Meiotic X-inactivation and sexual antagonism can only partly account for these patterns. This study shows that dosage compensation (DC) in Drosophila may contribute substantially to the depletion of male genes on the X. To equalize expression between X-linked and autosomal genes in the two sexes, male Drosophila hypertranscribe their single X, whereas female mammals silence one of their two X chromosomes. Fine-scale mapping data of dosage compensated regions was combined with genome-wide expression profiles to show that most male-biased genes on the D. melanogaster X are located outside dosage compensated regions. Additionally, X-linked genes that have newly acquired male-biased expression in D. melanogaster are less likely to be dosage compensated, and parental X-linked genes that gave rise to an autosomal male-biased retrocopy are more likely located within compensated regions. This suggests that DC contributes to the observed demasculinization of X chromosomes in Drosophila, both by limiting the emergence of male-biased expression patterns of existing X genes, and by contributing to gene trafficking of male genes off the X (Bachtrog, 2010).
This study found compelling evidence that dosage compensation influences patterns of sex-biased expression in Drosophila, and contributes to movement of male-biased genes off the X. This analysis suggests that the deficiency of male-biased genes on the Drosophila X does not simply reflect a lack of dosage compensation at some genes but instead can partly be accounted for by dosage compensation directly interfering with further upregulation of MSL-bound, already hypertranscribed X-linked genes in males. The X chromosome in male Drosophila is encumbered by the MSL complex and its chromatin structure is modified globally, which may limit subsequent transcription factor binding or chromatin remodeling, and thus inhibit further transcriptional activation. Indeed, direct interference between chromatin remodeling complexes and the dosage compensation machinery has been reported in Drosophila. Additionally, male-biased gene expression originates mainly by increasing transcription of nonbiased genes in males (rather than downregulation in females, and higher expression levels may be harder to achieve on an already hypertranscribed chromosome. High-expression male-biased genes are located less often on the X than low-expression male-biased genes. This is expected if limits in rates of transcription prevent the accumulation of male-biased genes on the X, given that such limitations are less likely to affect genes that are transcribed only at low levels (Bachtrog, 2010).
Not all organisms show a deficiency of male-biased genes on the X. In particular, mammalian X chromosomes are enriched for single-copy genesis genes that are expressed premeiosis, and multicopy testis genes showing postmeiotic expression. This difference in X-chromosomal gene content between taxa could result from fundamental differences in the mechanisms of dosage compensation. Dosage compensation in mammals is achieved by first doubling global expression levels of the X in both sexes, followed by inactivation of one X in females. The chromatin structure of the active X in mammals and baseline transcription rates of X-linked genes thus appear the same between the sexes (even though they might differ from average autosomal rates of transcription), therefore imposing no male-specific restrictions on the evolution of sex-biased expression patterns. Thus, the difference in X-chromosomal gene content between Drosophila and mammals -- with a deficiency versus an accumulation of male-biased genes -- may be understood in light of their vastly different dosage compensation mechanisms (Bachtrog, 2010).
Germ cells must develop along distinct male or female paths to produce the sperm or eggs required for sexual reproduction. In both mouse and Drosophila, sexual identity of germ cells is influenced by the sex of the surrounding somatic tissue, but little is known about how the soma controls germline sex determination. This study shows that the JAK/STAT pathway provides a sex-specific signal from the soma to the germline in the Drosophila embryonic gonad. The somatic gonad expresses a JAK/STAT ligand, unpaired (upd), in a male-specific manner, and activates the JAK/STAT pathway in male germ cells at the time of gonad formation. Furthermore, the JAK/STAT pathway is necessary for male-specific germ cell behavior during early gonad development, and is sufficient to activate aspects of male germ cell behavior in female germ cells. This work provides direct evidence that the JAK/STAT pathway mediates a key signal from the somatic gonad that regulates male germline sexual development (Wawersik, 2005).
While investigating communication between the somatic gonad and germline, the JAK/STAT pathway was found to be specifically activated in male, but not female, germ cells. In Drosophila, JAK/STAT signaling is initiated when an UPD or UPD-like ligand binds a transmembrane receptor (Domeless), activating the JAK Hopscotch (HOP), which phosphorylates the STAT92E transcription factor. STAT activation has been shown to regulate stat gene expression and can induce upregulation of the STAT92E protein, which can be used as an assay for JAK/STAT pathway activation. STAT92E is upregulated specifically in male, but not female germ cells at the time of gonad formation. This reflects male-specific activation of the JAK/STAT pathway since (1) the activated form of STAT92E (phospho-STAT92E) is also detected in only male germ cells, and (2) JAK activity is necessary and sufficient for STAT92E expression. Expression of a JAK inhibitor, Socs36E, results in loss of STAT92E expression in male germ cells and expression of constitutively active JAK (hopTumL) induces STAT92E in female germ cells. The male-specific activation of STAT92E at this time is distinct from STAT92E activation in germ cells in the early embryo, which is not sex-specific and is regulated by the MAP kinase pathway (Wawersik, 2005).
It was also found that STAT92E expression in male germ cells is dependent on their association with the somatic gonad. STAT92E is not detected in germ cells that are migrating to the gonad, but is detected in male germ cells after they contact the somatic gonad. STAT92E expression is greatly reduced or absent in eya mutants, where somatic gonad identity is initiated, but not maintained. Furthermore, STAT92E is not detected in germ cells found outside the gonad in wild type embryos or in mis-localized germ cells in wunen and HMG-CoA reductase mutants which lack guidance cues that target germ cells to the somatic gonad. However, in these same mutants, STAT92E is detected in the few germ cells that contact the somatic gonad in male embryos (Wawersik, 2005).
STAT92E expression in the germline is dependent on the sex of the surrounding soma. When XX (normally female) germ cells were present in a soma that was masculinized by expression of the male form of the somatic sex determination gene doublesex (dsx), germ cells now expressed STAT92E. dsx does not play an autonomous role in germ cells themselves, indicating that STAT92E induction in these embryos is caused by masculinization of the soma. Conversely, when the somatic gonad of an XY (normally male) embryo is feminized by expression of the sex determination gene transformer (tra) in the mesoderm, but not germ cells, STAT92E expression is no longer observed in XY germ cells. Taken together, these data indicate that the male somatic gonad is necessary and sufficient to activate the JAK/STAT pathway in either XX or XY germ cells (Wawersik, 2005).
Consistent with this, it was found that the JAK/STAT ligand, upd, is expressed specifically in the male, but not female, somatic gonad. Expression of STAT92E in male germ cells was no longer detected in embryos in which upd and two homologs, upd2 and upd3, are deleted [Df(os1a]. Since male germ cells from embryos mutant for upd alone still express STAT92E, JAK/STAT activation in the germline may be regulated redundantly by upd and one or more of its homologs. In addition, expression of upd in either the mesoderm or germ cells is sufficient to induce STAT92E expression in XX germ cells. Expression of upd2 or upd3 is also capable of inducing STAT92E in germ cells (Wawersik, 2005).
upd is also important for embryonic patterning and somatic sex determination. Interestingly, upd promotes female identity in the soma, but promotes male development in the germline. To verify that the effects of upd on the germline are not indirectly caused by other effects of upd, indicators of embryonic segmentation (Engrailed), somatic sex determination (Sex lethal), somatic gonad identity (Eyes absent), and somatic gonad sexual identity (Sox100B) were examined. Df(os1a) hemizygous male embryos exhibit segmentation defects as expected, but form gonads that express normal somatic and sex-specific markers. Embryos ectopically expressing upd are normal in all respects examined (Wawersik, 2005).
Whether activation of the JAK/STAT pathway by the male somatic gonad regulates male-specific development of germ cells was examined. In adult testes, the JAK/STAT pathway is required for maintenance of germline stem cells, making it difficult to assess the role of this pathway on male germ cell identity at this stage. Instead, germ cells were examined during embryogenesis and early larval stages, when germ cell development first becomes sexually dimorphic. In the mouse, the earliest manifestation of sex determination in the germline is differential regulation of the germline cell cycle by the soma. In Drosophila, germ cells undergo 1-2 divisions after their formation, but are arrested in the cell cycle during germ cell migration and only resume division shortly after the gonad has formed. Since larval testes contain more germ cells than larval ovaries, whether proliferation is regulated differently in male and female germ cells was examined. Indeed, sex-specific analysis of a mitotic marker (phosphohistone-H3) in the germline indicates that germ cell proliferation is entirely male-specific during early stages of gonad development. Furthermore, male-specific germ cell division is dependent on the male somatic gonad. Male germ cells do not proliferate in eya mutants that lack the somatic gonad, or in lost germ cells within wunen mutant embryos. XX germ cells in a masculinized soma (dsxD/ dsx1) proliferate, while XY germ cells in a feminized soma (UAS-traF; twist-Gal4) do not. Thus, the pattern of germ cell proliferation correlates exactly with activity of the JAK/STAT pathway in germ cells (Wawersik, 2005).
To assess whether JAK/STAT signaling regulates male-specific germ cell division, embryos lacking zygotic Stat92E activity were examined and a dramatic decrease was observed in male germ cell proliferation. Similar reductions in germ cell proliferation are observed in the upd/upd-like mutant (Df(os1a)) and in embryos where the JAK inhibitor Socs36E is expressed in germ cells. Thus, JAK/STAT activity is required within germ cells for proper male-specific germ cell division in the gonad. Expression of upd in the germline is sufficient to induce proliferation in female germ cells. Thus, the JAK/STAT pathway can induce XX germ cells to exhibit this male-specific germ cell behavior (Wawersik, 2005).
Whether the JAK/STAT pathway regulates other aspects of male germ cell development was examined. male germline marker-1 (mgm-1) is a lacZ enhancer trap line that is expressed in male germ cells, but not female germ cells, and therefore is a marker for male germ cell identity. Inhibiting the JAK/STAT pathway by removing zygotic Stat92E activity does not affect mgm-1 expression in the embryo, which is as expected since initial mgm-1 expression is dependent on germ cell autonomous cues. However, removal of zygotic Stat92E activity completely abolished mgm-1 expression in first instar larvae. In wild-type first instar male larvae, mgm-1 expression is observed in most germ cells, which are likely to be developing male germline stem cells and spermatogonia. No mgm-1 expression is observed in Stat92E-mutant larvae, and β-galactosidase expression is only observed in the soma, not the germline, in the pattern expected from the Stat92E P element allele. In an experiment where 25% of larvae were expected to be both male and contain the mgm-1 enhancer trap, 23.2% (n=262) of wild type larvae exhibited mgm-1 expression in the germ cells, while no Stat92E mutant larvae exhibited germ cell mgm-1 expression; this is significantly different from wild type siblings. Thus, Stat92E mutants exhibit a strong effect on male germline development, and some male germline cell types are either missing, or have an altered identity (Wawersik, 2005).
Finally, the extent to which activation of the JAK/STAT pathway can masculinize female germ cells was assessed. Female germ cells expressing upd are not expected to be fully masculinized because, although a male-specific signal is being activated, these germ cells are otherwise still in a female somatic environment and retain female germ cell autonomous cues. Indeed, such embryos give rise to fertile adult females, indicating that at least some germ cells retain, or revert back to, a female identity. This may be due, in part, to the failure of the upd construct to be expressed in the adult female germline. However, upd is sufficient to induce male-specific gene expression in embryonic XX germ cells. While mgm-1 is normally expressed only in germ cells in males, mgm-1 was expressed in all embryos when upd was ectopically expressed. In addition, two new male germline markers, disc proliferation abnormal (dpa) and minichromosome maintenance 5 (mcm5), were identified, that can also be induced by upd. Whereas these genes are normally expressed in germ cells only in males, female embryos exhibit germ cell expression of these genes when upd is ectopically expressed. In an experiment where only 50% of embryos are expected to express ectopic upd in the germline, 32.5% of female embryos expressed dpa and 21.3% expressed mcm5. Therefore, upd expression is sufficient to activate male-specific gene expression in female germ cells (Wawersik, 2005).
These data indicate that the JAK/STAT pathway mediates a critical signal from the male somatic gonad that is required for male germ cell development. This signal likely acts together with male germ cell autonomous cues to promote male germline identity and spermatogenesis. This signal is also sufficient to activate the male pattern of proliferation and gene expression in female germ cells, even when these germ cells retain female germ cell autonomous cues and are present in an otherwise female soma. It will be very interesting in the future to identify additional (e.g. female) somatic signals, along with germ cell autonomous cues, and to assess the relative contribution of these factors to proper germline sexual development. Since one of the earliest aspects of sex-specific germ cell behavior in both Drosophila and mouse is the regulation of the germline cell cycle by the somatic gonad, it will be of further interest to determine if the somatic signals operating in Drosophila play a similar role in germline sex determination in mammals (Wawersik, 2005).
Sex determination in Drosophila is commonly thought to be a cell-autonomous process, where each cell decides its own sexual fate based on its sex chromosome constitution (XX versus XY). This is in contrast to sex determination in mammals, which largely acts nonautonomously through cell-cell signaling. This study examined how sexual dimorphism is created in the Drosophila gonad by investigating the formation of the pigment cell precursors, a male-specific cell type in the embryonic gonad surrounding the testis. Surprisingly, sex determination in the pigment cell precursors, as well as the male-specific somatic gonadal precursors, was found to be non-cell autonomous. Male-specific expression of Wnt2 within the somatic gonad triggers pigment cell precursor formation from surrounding cells. These results indicate that nonautonomous sex determination is important for creating sexual dimorphism in the Drosophila gonad, similar to the manner in which sex-specific gonad formation is controlled in mammals (DeFalco, 2008).
This study has shown that two distinct male-specific cell types in the Drosophila gonad exhibit nonautonomous sex determination. For both the male specific somatic gonadal precursors (msSGPs) and the pigment cell (PC) precursors, the sex determination pathway does not act in these cells themselves, and both are dependent on sex-specific signaling from the SGPs in order to develop properly as male or female. These findings are in contrast to the commonly held view that sex determination in Drosophila is a cell-autonomous process, and demonstrate the similarity in sex-specific gonad development between flies and mammals (DeFalco, 2008).
This study has identified a novel, sex-specific cell type in the Drosophila embryonic gonad, the PC precursors, and studied the mechanism by which the sex determination switch controls the sex-specific development of these cells. The data indicate that male-specific expression of Wnt2 in the SGPs of the gonad signals nonautonomously to the fat body to form PC precursors. dsx ensures that PC formation is male-specific by repressing Wnt2 expression in female gonads in late-stage embryos (stage 17). The sex of the fat body itself does not affect PC precursor formation, since cells with a female identity can form PC precursors when associated with a male gonad or with a female gonad that expresses Wnt2. Furthermore, Wnt2 acts directly on the fat body, since blocking Wnt signaling in male fat body cells prevents them from forming PC precursors. PC precursor identity in the fat body is regulated by ems acting upstream of Sox100B in response to the Wnt2 signal. An interesting question is whether Wnt2 is a direct downstream target of DSX in controlling sexual dimorphism. The DNA binding specificity for DSX has been determined, and there are a number of sites upstream of the Wnt2 start of transcription that either exactly match or closely match the DSX binding consensus sequence. Several of these sites are conserved between different Drosophila species. However, a fragment of the Wnt2 promoter has not yet been identified that allows testing of whether Wnt2 expression in the somatic gonad is directly regulated by DSX, since the upstream region that includes the putative DSX binding sites does not promote expression in the gonad (DeFalco, 2008).
The creation of sexual dimorphism in the PC precursors differs from that of the msSGPs. While the PC precursors are apparently only specified in males and recruited to form part of the testis, msSGPs are initially specified in both sexes, and are only present in the male gonad because they undergo programmed cell death specifically in females. Furthermore, the germline stem cell niche in the testis (the hub) is formed from a population of anterior SGPs that are present in the gonads of both sexes, but only form the hub in males and presumably form part of the ovary in females. These events are all regulated by dsx, and demonstrate the diverse cellular mechanisms that a sex determination gene can utilize to control sexual dimorphism. Interestingly, in dsx null mutant embryos each of these cell types develops as if it were male. Thus, the male mode of development can at least be initiated in these cell types in the absence of dsx function, and dsx primarily acts in females to repress male development. dsx is clearly required in males at some point for proper testis formation, therefore some cell types in the gonad may not be entirely masculinized in dsx mutants (DeFalco, 2008).
The nonautonomous nature of PC precursor specification contrasts with the commonly held view that sex determination in Drosophila is a cell-autonomous process, where 'every cell decides for itself' whether it should develop as male or female based on its own intrinsic sex chromosome constitution. This study has shown that the msSGPs undergo nonautonomous sex determination. The data indicate that a male-specific survival signal coming from the SGPs allows the msSGPs to survive and join the male gonad, while they undergo apoptosis in females. Finally, it has been shown that nonautonomous sex determination in the germ cells requires a male-specific signal from the SGPs that acts through the JAK/STAT pathway. Thus, not only does non-cell autonomous sex determination occur in the Drosophila gonad, it appears to be the predominant mechanism of sex determination. Of the cell types tested so far, only the hub cells, which form from a subset of SGPs, appear to decide their sexual fate in an autonomous manner. The current model is that the SGPs determine their sex in a cell-autonomous manner, and then signal to other cell types in the gonad (PC precursors, msSGPs, and germ cells) to control the sex-specific development of these cells via nonautonomous sex determination (DeFalco, 2008).
Nonautonomous sex determination is not limited to the gonad. Other tissues have been shown to decide their sex through cell-cell signaling. In the genital imaginal disc, the sexual identity of a signaling center, the A/P organizer, largely determines whether the disc will develop in the male or female mode. This is controlled non-cell autonomously through Wingless and Decapentaplegic signaling. In addition, sex-specific migration of mesodermal cells into the male genital disc is regulated by male-specific expression of the Fibroblast Growth Factor Branchless in the genital disc. Finally, in the nervous system, male neurons can non-cell autonomously induce the formation of the male-specific muscle of Lawrence from female muscle precursors. Given the large number of tissues and cell types that undergo nonautonomous sex determination, it seems that the conventional view can be abandoned that sex determination in Drosophila is an obligatorily cell-autonomous process; while some cell types utilize a cell-autonomous mechanism, many cell types clearly do not (DeFalco, 2008).
One reason why sex determination has been traditionally thought of as a cell-autonomous process in Drosophila is due to its relationship with X chromosome dosage compensation. This is the process by which gene expression from the single X chromosome in males is regulated to match that from the two X chromosomes in females. Both sex determination and X chromosome dosage compensation are regulated by the number of X chromosomes, acting through the master control gene Sex lethal (Sxl). It is likely that most or all cells count their X chromosomes and use this information to control X chromosome dosage in a cell-autonomous manner. However, as discussed above, it is now clear that cells do not necessarily use this information to decide their sex. Consistent with this idea, the expression of dsx, a key regulator of sex determination downstream of Sxl, is surprisingly tissue-specific. Within the embryo, dsx is only expressed in the SGPs and msSGPs of the gonad. Thus, not all cells even express the machinery to translate their sex chromosome constitution into sexual identity, and it is therefore necessary that sex-specific development of many cell types be controlled nonautonomously (DeFalco, 2008).
The nonautonomous cell-cell interactions that control gonad sexual dimorphism in Drosophila show great similarity to sex-specific gonad development in other species. In mammals, somatic sex determination is based on the presence or absence of the Y chromosome. The critical Y chromosome gene Sry is mainly expressed in a subset of cells in the somatic gonad in the mouse embryo, similar to dsx expression in the Drosophila embryonic gonad. Sry is only thought to be important for formation of Sertoli cells in males, and the sexually dimorphic development of all other cell types is thought to be regulated by local cell-cell interaction or hormonal cues. An excellent example of nonautonomous sex determination in the mouse is the recruitment of cells from the neighboring mesoderm (mesonephros) to form specific cell types in the mouse testis. Recruitment of these cells is dependent on the sex of the gonad, not the sex of the mesonephros. In addition, sex-specific development of other somatic cell types in the mouse gonad is regulated nonautonomously by cell-cell interaction, as is sexual identity in the germline. Thus, the use of non-cell autonomous sex determination and sex-specific cell recruitment are common mechanisms for creating gonad sexual dimorphism in flies and mice (DeFalco, 2008).
Nonautonomous sex determination in the mouse also utilizes signaling through the Wnt pathway. Wnt4 acts as a 'pro-female' gene that opposes Fibroblast growth factor 9 to regulate sex determination in the gonad. In early stages of gonad development, Wnt4 knockout females form a male-specific coelomic blood vessel and exhibit ectopic migratory steroidogenic cells, suggesting that Wnt4 acts to inhibit endothelial cell and steroid cell migration from the mesonephros into the female gonad. Interestingly, Wnt4 also has been shown to have a role in the male gonad, as male knockout mice show defects in Sertoli cell differentiation, downstream of Sry but upstream of Sox9. Wnt7a also has been implicated in sexual dimorphism in the reproductive tract, as Wnt7a knockout mice fail to express Mullerian-inhibiting substance (MIS) type II receptor in the Mullerian duct mesenchyme, which is required for regression of the duct in male embryos. In addition, a number of Wnt genes have been found to be expressed sex-specifically in the gonad through sex-specific gene profiling, indicating that other Wnt family members play a role in creating sexual dimorphism in the mammalian gonad (DeFalco, 2008).
It is also interesting that several conserved transcription factors act during gonad development in diverse species. Sox100B is the fly homolog of SOX9/Sox9, a critical regulator of sex determination and male gonad development in humans and mice. Similarly, a mouse homolog of ems, Emx2, is expressed in the developing gonad and is required for development of the urogenital system. Lastly, dsx homologs of the DMRT family have been implicated in sex-specific gonad development in diverse species. Thus, not only are the cellular mechanisms, such as non-cell autonomous sex determination and cell-cell recruitment, common between flies and mice, but the specific genes that regulate sexually dimorphic gonad development may also be conserved. Since the formation of testes versus ovaries, and sperm versus egg, are critical features of sexual reproduction, they may represent processes that are highly conserved across the animal kingdom (DeFalco, 2008).
Social interactions depend on individuals recognizing each other, and in this context many organisms use chemical signals to indicate species and sex. Cuticular hydrocarbon signals are used by insects, including Drosophila, to distinguish conspecific individuals from others. These chemicals also contribute to intraspecific courtship and mating interactions. However, the possibility that sex and species identification are linked by common chemical signalling mechanisms has not been formally tested. This study provides direct evidence that a single compound is used to communicate female identity among flies, and to define a reproductive isolation barrier between Drosophila melanogaster and sibling species. A transgenic manipulation eliminated cuticular hydrocarbons by ablating the oenocytes (see Insect oenocytes: a model system for studying cell-fate
specification by Hox genes), specialized cells required for the expression of these chemical signals. The resulting oenocyte-less (oe-) females elicited the normal repertoire of courtship behaviours from males, but were actually preferred over wild-type females by courting males. In addition, wild-type males attempted to copulate with oe- males. Thus, flies lacking hydrocarbons are a sexual hyperstimulus. Treatment of virgin females with the aversive male pheromone cis-vaccenyl acetate (cVA) significantly delayed mating of oe- females compared to wild-type females. This difference was eliminated when oe- females were treated with a blend of cVA and the female aphrodisiac (7Z,11Z)-heptacosadiene (7,11-HD), showing that female aphrodisiac compounds can attenuate the effects of male aversive pheromones. 7,11-HD also was shown to have a crucial role in heterospecific encounters. Specifically, the species barrier was lost because males of other Drosophila species courted oe- D. melanogaster females, and D. simulans males consistently mated with them. Treatment of oe(-) females with 7,11-HD restored the species barrier, showing that a single compound can confer species identity. These results identify a common mechanism for sexual and species recognition regulated by cuticular hydrocarbons (Billeter, 2009).
D. melanogaster produces hydrocarbons of various chain lengths, including unbranched alkanes, methyl-branched alkanes, alkenes and derivatives thereof. The alkenes are expressed sex-specifically, and have been associated with both sex and species discrimination. Compared to females, males express high levels of the monoalkene (Z)-7-tricosene (7-T), which has been reported to increase females' receptivity to mating attempts. Moreover, 7-T is repulsive to other males and may prevent male-male interactions. In contrast, females produce sex-specific dienes such as (7Z,11Z)-heptacosadiene (7,11-HD) and (7Z,11Z)-nonacosadiene (7,11-ND), which act as aphrodisiac pheromones for D. melanogaster males. Hydrocarbons are strongly associated with sexual recognition, because wild-type males court males that have been genetically modified to express female hydrocarbons, indicating that the mutants are perceived as females hydrocarbons (Billeter, 2009).
There are still large gaps in knowledge of the functions of individual hydrocarbons and the tissues where these compounds are synthesized. As in other insects, specialized cells called oenocytes, located on the inner surface of the abdominal cuticle, are thought to be the site of hydrocarbon biosynthesis in D. melanogaster. Consistent with this hypothesis, desaturase 1 (desat1), which encodes an enzyme involved in hydrocarbon synthesis, is expressed in Drosophila oenocytes (Marcillac, 2005). Previous studies have demonstrated that genetic feminization of these cells results in production of female hydrocarbons by male flies; however, these and other manipulations have been confounded by the concurrent feminization of cells in many other sexually dimorphic tissues, including the central nervous system. To test the hypothesis that these cells are required for production of chemical signals used in sexual and species recognition, the Gal4-UAS system was used to target transgene expression specifically to the adult oenocytes. An oenocyte Gal4 driver was generated, derived from the regulatory sequence of one of the desat1 promoters (Marcillac, 2005) that is expressed specifically in oenocytes of adult females. The driver is also expressed in the larval oenocytes and in the reproductive organs of adult males. This driver was used to ablate adult oenocytes by inducing expression of the pro-apoptotic gene head involution defective (hid). This approach initially caused lethality in larvae, probably due to the destruction of the larval oenocytes. To circumvent this problem blocked the driver's action was blocked during development using the Tubulin-Gal80ts transgene. Using this method, flies were generated without oenocytes (oe-). Analysis of whole-body hydrocarbon extracts confirmed that both oe- males and females were essentially devoid of these compounds, showing that the oenocytes are necessary for hydrocarbon display in D. melanogaster. The male pheromone cis-vaccenyl acetate (cVA) was unaffected in oe- males because this compound is synthesized in the ejaculatory bulb. The oe- transgenic strain therefore provided a 'blank slate' for evaluating the role of hydrocarbons in intra- and interspecific communication hydrocarbons (Billeter, 2009).
Sexual behaviour of oe- flies was assayed to test hydrocarbon function during reproduction. Despite the association of hydrocarbon signals and Drosophila courtship, absence of these signals did not alter courtship behaviours per se. The oe- males displayed normal courtship behaviour towards wild-type females, but slightly less intense than control males. However, wild-type females were less receptive to oe- males than control males, with oe- males taking almost four times as long to achieve mating. Thus, hydrocarbons of males do not seem to affect their own courtship behaviour, but rather, influence the receptivity of females to their mating attempts. However, the influence of non-oenocyte cells within the male reproductive organs that may have been affected by the ablation cannot be excluded. Notably, oe- males elicited courtship and copulation attempts from both wild-type males and other oe- males, indicating that oe- males were perceived as females, even though all other male characteristics were present. The vigorous courtship of oe- males by each other resulted in unnatural behaviours such as engaging one another by rotating in a head-to-head orientation, and males attempting copulation with each other's heads. These behaviours were suppressed by treatment of oe- males with synthetic 7-T, confirming the function of 7-T in inhibiting male-male interactions hydrocarbons (Billeter, 2009).
Wild-type males exhibited normal courtship behaviour towards oe- females, apparently undeterred by the lack of female . However, mating latency was significantly shorter, and when given a choice between an oe- and a control female, wild-type males preferred oe- females. Together, these data indicate that females lacking hydrocarbons are more attractive than those with a normal hydrocarbon profile. This suggests that female hydrocarbons normally act to slow down male mating attempts, facilitating assessment of a potential partner's species and fitness. Thus, any oe- fly, irrespective of its development as female or male, seems to sexually hyperstimulate males. It is hypothesized that hydrocarbons normally act to superimpose sexual identity on an otherwise attractive fly substrate hydrocarbons (Billeter, 2009).
The results described above suggested that female attractiveness depends on a balance between attraction/stimulation and repulsion/deterrence. This was investigated by treating females with the aphrodisiac compound 7,11-HD, and with cVA, which males transfer to females via the ejaculate to deter further mating attempts by other males. Whereas cVA decreases the probability that females will re-mate, wild-caught females produce offspring from multiple sires, indicating that polyandry is common and that the effect of cVA is not absolute. O- flies were treated with doses of these compounds approximating wild-type levels. The mating latency of wild-type males with oe- females treated with 7,11-HD was not different from that with untreated oe- females, indicating that 7,11-HD alone does not affect attractiveness of oe- females. As expected, treating wild-type females with increasing doses of cVA delayed mating accordingly, and the effect was even more pronounced with oe- females treated with the same doses of cVA. This effect was not due to differences in the rates of release of cVA from the control and oe- flies, as shown by the profiles of cumulative loss of cVA over time for the two genotypes. Instead, the exaggerated effect of cVA on oe- females is consistent with the hypothesis that the aversive effects of this compound are normally moderated by the presence of other hydrocarbons. Indeed, when cVA and 7,11-HD were applied together, the mating latencies of oe- and wild-type females were indistinguishable. Apparently, 7,11-HD mitigated the deterrent effects of cVA. The data suggest that a male's perception of a female's availability is normally regulated by a mixture of attractive and aversive signals. From an evolutionary perspective, the combined effect of a female attractant with a male deterrent may illustrate an instance of post-copulatory sexual conflict in which the attractant solicits additional mates despite the first male's effort to render a female unattractive by marking her with cVA hydrocarbons (Billeter, 2009).
In addition to mediating conspecific reproductive interactions, the hydrocarbons of female D. melanogaster have an important role in reproductive isolation between species. For example, within the nine species of the melanogaster subgroup, only D. melanogaster, D. sechellia and D. erecta produce female-specific dienes. Females in the rest of the subgroup express the same hydrocarbons as males. Males of species with non-sexually dimorphic hydrocarbons generally do not court females from dimorphic species, indicating that the dienes might act as reproductive isolation barriers between these species groups. Furthermore, males from dimorphic species do not vigorously court females from non-dimorphic species. In contrast, males of all species in the melanogaster subgroup have similar hydrocarbons, including abundant 7-T. Finally, D. melanogaster females lacking hydrocarbons are courted by at least two sibling species, D. simulans and D. mauritiana. The behaviour of males from other species in the melanogaster subgroup towards oe- females was tested, to assess the contribution of oenocytes and hydrocarbons to reproductive isolating mechanisms. D. simulans and D. yakuba were tested as test species because they represent species in which the females lack dienes. D. erecta was included because it differs from D. melanogaster in the pattern of dienes expressed hydrocarbons (Billeter, 2009).
Males of all three species courted oe- D. melanogaster females, but exhibited limited or no courtship towards control D. melanogaster females. This indicates that oenocytes and their hydrocarbon products are major components of the reproductive isolation barrier, ensuring that courtship and mating attempts are only initiated between conspecifics. It has been proposed that 7,11-HD functions to create this barrier in D. melanogaster. To test this directly, oe- D. melanogaster females and wild-type females from the different species were treated with synthetic 7,11-HD. Treatment suppressed courtship by males of all three species, demonstrating that 7,11-HD alone is sufficient to create a species barrier. Interestingly, D. erecta males were blocked by 7,11-HD, despite the fact that hydrocarbons of D. erecta females include other dienes in common with those of D. melanogaster. Furthermore, D. melanogaster males actively courted D. erecta females, possibly because the diene 7,11-ND is also expressed by D. melanogaster females. D. simulans and D. yakuba females treated with 7,11-HD elicited strong courtship from D. melanogaster males. These results demonstrate the multifunctional role of 7,11-HD as an attractant and/or stimulant for some species and as a deterrent for others hydrocarbons (Billeter, 2009).
Despite attempting copulation, D. erecta males never mated with oe- females, suggesting that signals other than hydrocarbons are required to induce receptivity in these females. However, within a 24-h period, nearly all oe- D. melanogaster females mated with D. simulans males, whereas no control D. melanogaster females mated with these males. Treatment of oe- females with 7,11-HD completely blocked interspecific mating with D. simulans males, even at a dose five times lower than the amount found in females of wild-type D. melanogaster strain. Similar treatment of D. simulans females with 7,11-HD only delayed mating by D. simulans males. It is hypothesized that 7-T counters the effect of 7,11-HD in D. simulans females. This is because 7-T functions as an aphrodisiac for D. simulans males and is expressed in higher quantities in D. simulans females than in D. melanogaster females. D. simulans males were assayed with oe- females treated with either 7-T alone, or in combination with 7,11-HD. Synthetic 7-T alone induced a slight decrease in mating latency, indicating that 7-T is an attractant for D. simulans males. However, the striking effect of 7-T was to reduce the effect of 7,11-HD in a dose-dependent manner. These data parallel the balancing effect of 7,11-HD on cVA for D. melanogaster males. Thus, this study has demonstrated that female hydrocarbons orchestrate mating both within and between the species. Whereas a single compound such as 7,11-HD may be enough to establish a species barrier, the effect of this compound is moderated by the relative quantity of other signals. Indeed, the effects of 7,11-HD are particularly noteworthy because it functions as an attractant in an intraspecific context, whereas in an interspecific context, it aids in species recognition, thereby placing social communication and speciation on the same continuum hydrocarbons (Billeter, 2009).
The logic of pheromonal communication in Drosophila seems to be based on a foundation that imparts general attractiveness to a fly. In this study female oenocytes are the primary organ for communicating species and sex identity to males. Others have shown that males use species-specific acoustic tags within their love song for females during courtship. Thus, both acoustic and pheromonal tags establish a context for social interactions by regulating sex and species recognition. Given that individual flies regulate their own hydrocarbon display in accord with their social surroundings, it is plausible that these compounds also function in individual recognition hydrocarbons (Billeter, 2009).
The establishment of sexual identity is a crucial step of germ cell development in sexually reproducing organisms. Sex determination in the germline is controlled differently than in the soma, and often depends on communication from the soma. To investigate how sexual identity is established in the Drosophila germline, a molecular screen was conducted for genes expressed in a sex-specific manner in embryonic germ cells. Sex-specific expression of these genes is initiated at the time of gonad formation (stage 15), indicating that sexual identity in the germline is established by this time. Experiments where the sex of the soma was altered relative to that of the germline (by manipulating transformer) reveal a dominant role for the soma in regulating initial germline sexual identity. Germ cells largely take on the sex of the surrounding soma, although the sex chromosome constitution of the germ cells still plays some role at this time. The male soma signals to the germline through the JAK/STAT pathway, while the nature of the signal from the female soma remains unknown. The genes ovo and ovarian tumor (otu) are expressed in a female-specific manner in embryonic germ cells, consistent with their role in promoting female germline identity. However, removing the function of ovo and otu, or reducing germline function of Sex lethal, had little effect on establishment of germline sexual identity. This is consistent with findings that signals from the soma are dominant over germline autonomous cues at the initial stage of germline sex determination (Casper, 2009).
This analysis demonstrates that a sex-specific program of gene expression is present in the germline soon after the time of gonad formation [stage 15, ~12 hours after egg laying (AEL)]. Examples were identified of both male-specific germline genes and female-specific germline genes, with other genes being expressed equally in the two sexes. Therefore, it is likely that the sex-specific pattern of gene expression in the germline represents the establishment of true sexual identity in the germline, as opposed to other differences that might be observed in the germline between the sexes, such as proliferation status or transcriptional competence. It is concluded that sexual identity in the germline is established at least as early as stage 15. The observation that many genes examined initiate sex-specific germline expression at this time might indicate that this is when germline sexual identity is first established. However, it was also found that the soma signals to the germline in a sex-specific manner just prior to gonad formation (stage 13, ~10 hours AEL), indicating that germ cells could determine their sex even earlier. Because the zygotic genome is only robustly activated in the germline during gastrulation (stage 9, ~4 hours AEL), this sets a narrow time window during which sexual identity is established in the germline (Casper, 2009).
It is significant that germline sexual identity appears to be first manifested as the germ cells contact the somatic gonad. This is also the time that sexual dimorphism is first observed in the somatic gonad and that it exhibits sex-specific patterns of gene expression, Jheh2 and CG5149). The finding that germ cells might establish their sexual identity only as they contact the somatic gonad is consistent with the strong role of the soma in determining germline sexual identity. In addition, signaling from the germline back to the soma also occurs at this time (Kitadate, 2007; Casper, 2009).
The establishment of a sex-specific pattern of gene expression in the germline requires these cells to acquire germ cell identity, in addition to sexual identity. Previous work has established that germ cell-specific transcription is independent of the somatic environment, and is autonomously regulated in the germ cells. Indeed, it was seen that germ cell expression of the genes reported in this study can be independent of the somatic gonad. Germ cell-specific gene expression is likely to be regulated by the germ plasm, and several maternally expressed germline transcription factors have recently been identified (Yatsu, 2008; Casper, 2009).
Previously, it was known that sexual identity in the germline requires autonomous cues, along with non-autonomous cues from the surrounding soma, but little was known about how the signals work together to establish sexual identity. XX germ cells cannot develop normally in a male soma, and XY germ cells cannot develop normally in a female soma. Although evidence was found for both autonomous and non-autonomous regulation of germline sexual identity in the embryo, it appears that non-autonomous signals from the soma are dominant over germline autonomous cues. When XX germ cells are present in a male soma (tra mutants), they exhibit a clear increase in expression of male germ cell genes, and decreased expression of the female-specific gene otu. Similarly, XY germ cells present in a female soma (UAS-tra) exhibit a strong repression of male germ cell genes, while otu expression is increased. Thus, in each case the germ cells largely take on the sexual identity of the surrounding soma, independent of their own sex chromosome constitution. However, subtle differences in gene expression between XX and XY germ cells remain in these situations, indicating some germ cell autonomous control of sexual identity. In addition, when germ cells are present outside of the gonad (srp mutants), some differences are also observed between XX and XY germ cells. When outside of the gonad, XY germ cells are more likely than XX germ cells to express male-specific genes, and XX germ cells maintain some otu expression, while otu is largely off in XY germ cells. Thus, there is at least some autonomous contribution of the germ cell sex chromosome genotype to germline sexual identity at this early stage (Casper, 2009).
The genes of the ovarian tumor loci, ovo, otu, stil and Sxl, are thought to contribute to autonomous germline sexual identity by promoting female identity in XX germ cells. At this early stage, no change was observed in sex-specific germ cell gene expression in mutants for ovo, otu or stil, or in embryos with mutations that reduce the function of Sxl in the germline. Although this could indicate that these genes do not play a role in the initial establishment of germline sexual identity, these observations could also be due to the dominant effects of the soma on sex-specific germ cell gene expression at this time. These mutations would be expected to masculinize XX germ cells, causing them to exhibit a more male-like pattern of gene expression. However, even fully male (XY) germ cells exhibit a female pattern of gene expression when in a female soma. Thus, the dominant effect of the female soma might mask any masculinizing effects from the removal of ovarian tumor locus genes at this time (Casper, 2009).
Evidence is seen for at least two types of non-autonomous regulation of germline sexual identity by the soma, one coming from the male soma and the other from the female soma. Both XY and XX germ cells can activate male-specific gene expression when not in contact with the somatic gonad, but expression of these genes does not appear to be as robust as when germ cells are in contact with a male soma. Furthermore, when XX germ cells are in contact with a male soma, expression of the female germ cell gene otu is partially repressed. Thus, the male soma is required for full levels of male gene expression in the germ cells, and for the repression of female genes (Casper, 2009).
The signal from the male soma to the germline appears to be primarily, and perhaps exclusively, acting through the JAK/STAT pathway. Previously, it was shown that the male soma activates the JAK/STAT pathway in the germ cells, and that this is required for proper male germ cell behavior (Wawersik, 2005). This pathway is necessary for the proper male-specific proliferation of embryonic germ cells, and for the maintenance, but not the initiation, of male-specific mgm-1 expression (male germline marker-1, which appears to be an enhancer trap in the escargot locus). The JAK/STAT ligand UPD is also sufficient for the induction of germ cell proliferation and mgm-1 expression, and for partial induction of mcm5 and dpa expression. This study found that, when the JAK/STAT pathway is inactivated, sex-specific gene expression in the germline resembles that of germ cells that are not in contact with the somatic gonad. Ectopic expression of upd in females is able to induce some aspects of male-specific gene expression in XX germ cells, but not as robustly as when XX germ cells are present in a male soma. This is likely to be due to the fact that, when upd is expressed in an otherwise female soma, it is in competition with female signals that repress male gene expression. It is concluded that the JAK/STAT pathway is important for the regulation of germline sex determination by the male soma, and may be the primary or only signal from the male soma to the embryonic germ cells. It is essential for the male-specific pattern of germ cell proliferation, and for the robust initiation and maintenance of male-specific gene expression (Casper, 2009).
It is clear that the female soma also plays a key role in regulating the sexual identity of the germline. Both XX and XY germ cells exhibit some level of male-specific gene expression when outside of the gonad, but this expression is dramatically repressed when in contact with a female soma. This repression has also been observed with mgm-1. In addition, XY germ cells exhibit some female-specific otu expression when in contact with a female soma. Therefore, the female soma is essential for the proper sex-specific regulation of germline gene expression, although the nature of the signal from the female soma to the germ cells remains unknown (Casper, 2009).
The source of both the male and female somatic signals to the germline is likely to be the somatic gonad. The germ cells show signs of receiving the JAK/STAT signal only when in contact with male somatic gonad, and germ cells outside of the gonad do not appear to receive the proper sex-specific signals (e.g. srp mutants). Furthermore, it is known that somatic regulation of germline sex is controlled by the sex determination cascade, acting through tra and dsx. However, the only cells to express DSX within the embryo are part of the somatic gonad. Thus, the somatic control over germline sex determination is likely to represent local signaling within the gonad environment, rather than long-range signaling from other somatic cell types (Casper, 2009).
Somatic control over germline sex determination is a common feature of germ cell sexual development. In the mouse, germ cells first manifest sex-specific behaviors as they contact the somatic gonad in the genital ridge. The initial behavior of the germ cells is dependent on the sex of the soma, as male germ cells exhibit female behavior (early meiosis) when contacting a female soma, and female germ cells exhibit male behavior when in contact with a male soma. At least some aspects of this sex-specific behavior are regulated by retinoic acid (RA) levels controlled by the soma; female germ cells receive a high level of RA signal, but the male soma degrades RA so that male germ cells receive less of this signal. Interestingly, this study found that the somatic gonad in Drosophila expresses Jheh2 in a male-specific manner. JHEH2 hydrolyzes Juvenile Hormone (JH), which is structurally related to RA. JH analogs have also been observed to influence sex determination in crustaceans, which further supports this interesting parallel between vertebrates and invertebrates (Casper, 2009).
In mammals, the germline sex chromosome constitution is also important for germ cell development. As in Drosophila, mouse and human Y chromosome genes are crucial for spermatogenesis. However, the number of X chromosomes also appears to play an autonomous role in germline sexual development in mammals. XX germ cells in a male soma [e.g. in Sex reversed (Sxr) mice appear initially male and do not enter meiosis, but eventually die. XO germ cells in a male soma survive and progress further in spermatogenesis. Similarly, having two X chromosomes promotes female germ cell identity. XX germ cells are predisposed to enter meiosis on the female timetable compared with XO germ cells under the same conditions, and are biased towards a female pattern of imprinting. Thus, as in Drosophila, the germ cell genotype contributes to germ cell sex determination in the mouse and depends on the number of X chromosomes. Similarly, humans with altered sex chromosome constitutions, such as those with Turner's Syndrome (XO females) or Klinefelter's Syndrome (XXY males) have relatively normal somatic development but exhibit severe germline defects, indicating that the proper number of X chromosomes in the germline is essential for germline development (Casper, 2009).
However, in other species, the sex chromosome constitution of germ cells does not play an important role in germline sex determination. In the housefly, Musca domestica, transplanted germ cells develop normally according to the somatic sex of their host and produce fertile gametes. Some species also exhibit dramatic sexual plasticity in the germline, with the same individual being able to produce both sperm and egg, such as in hermaphrodites (e.g., C. elegans or in species that exhibit natural sex reversal. i.e., wrasses and gobies). Given the diversity of mechanisms animals use for establishing sex determination in the soma, it is not surprising that there might be considerable diversity in the germline as well. However, it appears that an important role for the soma in controlling sexual identity in the germline is a common theme in germ cell development. Whether or not the sex chromosome constitution of the germline is also important for fertility in an organism places additional constraints on the evolution of sex determination mechanisms, sexual plasticity and the sex chromosomes (Casper, 2009).
Sex determination in the Drosophila germ line is regulated by both the sex of the surrounding soma and cell-autonomous cues. How primordial germ cells (PGCs) initiate sexual development via cell-autonomous mechanisms is unclear. This study demonstrates that, in Drosophila, the Sex lethal (Sxl) gene acts autonomously in PGCs to induce female development. Sxl is transiently expressed in PGCs during their migration to the gonads; this expression, which was detected only in XX PGCs, is necessary for PGCs to assume a female fate. Ectopic expression of Sxl in XY PGCs was sufficient to induce them to enter oogenesis and produce functional eggs when transplanted into an XX host. These data provide powerful evidence that Sxl initiates female germline fate during sexual development (Hashiyama, 2011).
Primordial germ cells (PGCs) are able to differentiate into eggs or sperm. It is thought that PGCs do not assume a sexual fate until they reach the gonads, where sexual dimorphism is imposed by both the sex of the surrounding soma and cell-autonomous cues. In Drosophila, pole cells or PGCs differentiate to a male fate in response to JAK/STAT signaling from the gonadal soma. The method by which female sexual development is initiated in pole cells, however, has not been elucidated. To clarify the mechanism that initiates a female fate in pole cells, a female-specific marker for this cell type was identified. Although several sex-specific markers, including mgm-1, disc proliferation abnormal, and minichromosome maintenance 5, have been reported, they are all expressed only in male pole cells after gonad formation (stage 15), based on signals from the male gonadal soma. lesswright (lwr), a gene that regulates posttranslational modification of proteins by small ubiquitin-related modifiers, is expressed in pole cells during embryogenesis. lwr is not characterized by sex-specific expression. When a dominant-negative form of lwr (lwrDN) was expressed in the pole cells of either sex, however, apoptosis was induced only in female (XX) pole cells during migration to the gonads. This effect caused a significant reduction in the number of XX pole cells in the gonads. Introduction of female-specific germline apoptosis induced by a dominant-negative form of lwr (f-gal) provides a previously uncharacterized marker of female sexual identity in migrating pole cells (Hashiyama, 2011).
Sex determination is controlled by the Sex lethal (Sxl) gene, which is first expressed at the blastodermal stage in the embryonic soma. Sxl encodes an RNA binding protein involved in alternative splicing and translation. In the soma of XX embryos, it functions through transformer (tra) and transformer-2 (tra-2), which in turn regulate alternative splicing of the doublesex (dsx) gene to produce a female-specific form of Dsx. In male (XY) embryos, this pathway is turned off, and a male-specific form of Dsx is produced by default. These Dsx proteins determine the sexual identity of somatic tissues. Previous reports, however, suggested that Sxl does not induce female sexual development in the germ line, as it does in the soma. Although Sxl is autonomously required for female sexual development, constitutive mutations in Sxl (SxlM) that cause XY animals to undergo sexual transformation from male to female do not necessarily interfere with male germline development. Moreover, tra, tra-2, and dsx are not required for female germline development. Finally, female-specific Sxl expression has been detected later in gametogenesis, but not in early germline development (Hashiyama, 2011).
Contrary to previous observations, this study found that Sxl was expressed in XX but not XY pole cells during their migration to the gonads. In the soma, Sxl transcripts are first expressed from the establishment promoter (Sxl-Pe) in a female-specific manner. Using a probe specific to the early transcript derived from Sxl-Pe, in situ hybridization signals were detected in migrating XX pole cells at around stage 9/10. Transgenic embryos, which expressed enhanced green fluorescent protein (EGFP) under the control of the Sxl-Pe promoter, were used to further confirm this female-specific Sxl-Pe activation. RTPE and sequencing analyses in pole cells were used to detect early Sxl transcripts that had the same sequence as the transcripts expressed in the soma (Hashiyama, 2011).
Next, it was determined whether Sxl feminize early pole cells using f-gal as a marker for female identity. It was found that the loss-of-function mutation SxlfP7B0 represses f-gal in XX pole cells. This repression is unlikely to result from sexual transformation of the soma, because an amorphic tra-2 mutation, which alters somatic sex, did not affect f-gal. Conversely, when the expression of Sxl together with lwrDN is forced in pole cells from stage 9 onward by using nanos-Gal4 and UAS-Sxl, f-gal is ectopically observed in XY pole cells. Sxl alone does not induce apoptosis or developmental defects in pole cells. These observations suggest that female sexual identity of migrating pole cells is regulated cell-autonomously by Sxl (Hashiyama, 2011).
It was then determined whether Sxl induces female development in XY pole cells. Because XY soma produces signals that direct XX germline cells to a male fate, XY pole cells expressing Sxl were transplanted into XX females, and their developmental fate was examined. Even in the presence of a gain-of-function Sxl mutation (SxlM1) that causes XY soma to transform from male to female, XY (or XO) pole cells enter the spermatogenic pathway when transplanted into XX females. These results suggest that Sxl is not sufficient to activate female germline development. SxlM1 mutations, however, do not affect transcription from the Sxl-Pe promoter, but instead structurally alter the late transcript from the Sxl maintenance promoter (Sxl-Pm), which allows Sxl protein production in both males and females. Consistent with this observation, Sxl transcripts derived from Sxl-Pe were detected in the pole cells of only female SxlM1 embryos. Thus, the SxlM1 mutation does not result in Sxl expression in XY pole cells as early as in XX pole cells (Hashiyama, 2011).
Instead, nanos-Gal4 and UAS-Sxl were used to induce Sxl expression in XY pole cells. Three types of XY pole cells were transplanted, each characterized by a different duration of Sxl expression: (1) XY pole cells in which Sxl was expressed from stage 9 until stage 16/17 using maternal nanos-Gal4 (XY-mSxl), (2) XY pole cells in which Sxl was expressed from stage 15/16 onward using zygotic nanos-Gal4 (XY-zSxl), and (3) XY pole cells in which Sxl was expressed from stage 9 onward using both maternal and zygotic nanos-Gal4 (XY-mzSxl). XY-mzSxl and XY-mSxl pole cells entered the oogenic pathway and produced mature oocytes in XX females. These oocytes contributed to progeny production. Thus, the XY pole cells produced functional eggs, even though oogenesis and egg production were reduced compared with XX pole cells. In contrast, XY-zSxl pole cells did not enter the oogenic pathway in almost all (92.3%) of the XX female hosts and instead were characterized by a tumorous phenotype, an indication of XY germline cells that have maintained male characteristics. Control XY pole cells from the embryos expressing Sxl only in the soma (XY-nullo-Sxl) showed a similar phenotype to that of XY-zSxl pole cells. These observations demonstrate that Sxl expression in XY pole cells during embryogenesis induces functional egg differentiation in the female soma (Hashiyama, 2011).
Sxl-specific double-stranded RNA (UAS-SxlRNAi) under the control of maternal nanos-Gal4 was used to reduce Sxl activity in XX pole cells during embryogenesis. Introducing UAS-SxlRNAi resulted in tumorous and agametic phenotypes in female adults, indicating that the XX germ line lost female characteristics. Taken together, these results show that Sxl acts as a master gene necessary and sufficient to induce female development in pole cells (Hashiyama, 2011).
XY-mzSxl pole cells adopted a male fate and executed spermatogenesis when they developed in an XY male soma. This observation suggests that the male soma plays a dominant role in determining the male germline fate, overriding the feminizing effect of Sxl. Another possibility is that the XX female soma plays a critical role in maintaining the Sxl-initiated female germline fate. Indeed, an XX germ line in the male soma shows a male gene-expression profile, whereas an XY germ line in the female soma exhibits a female expression profile, although these germ lines does not execute gametogenesis. Thus, female germline development requires interactions between the germline and somatic cells, in addition to germline-autonomous mechanisms involving Sxl (Hashiyama, 2011).
In mice, germline sexual identity is also regulated by both germline-autonomous and somatic signals. In the coelenterate Hydra, the germline sex is not influenced by the surrounding soma, and the germ line determines the phenotypic sex of the polyp. Thus, germline-autonomous regulation of sex has probably been present throughout the evolution of animals, and somatic control may have evolved with the emergence of mesodermal tissues, including gonadal soma. Sxl does not appear to play a key role in sex determination in non-drosophilid animals. Nevertheless, future studies should determine whether Sxl homologs are expressed in the germ line of non-drosophilids. Moreover, it would be of particular interest to identify downstream targets of Sxl in the Drosophila germ line and to test whether these genes have a widespread role in germline sex determination (Hashiyama, 2011).
Establishing germ cell sexual identity is critical for development of male and female germline stem cells (GSCs) and production of sperm or eggs. Germ cells depend on signals from the somatic gonad to determine sex, but in organisms such as flies, mice, and humans, the sex chromosome genotype of the germ cells is also important for germline sexual development. How somatic signals and germ-cell-intrinsic cues combine to regulate germline sex determination is thus a key question. This study found that JAK/STAT signaling in the GSC niche promotes male identity in germ cells, in part by activating the chromatin reader Phf7. Further, it was found that JAK/STAT signaling is blocked in XX (female) germ cells through the action of the sex determination gene Sex lethal to preserve female identity. Thus, an important function of germline sexual identity is to control how GSCs respond to signals in their niche environment (Bhaskar, 2022).
This study presents data that provides new insights into germline sex determination and the regulation of male versus female GSC identity. First, it was found that one key function of the JAK/STAT pathway in GSCs is to promote male identity and directly activate expression of the male germline chromatin regulator Phf7. Further, it was found that an important role for Sxl in female germ cells is to block the JAK/STAT pathway and prevent this signal from masculinizing the germline. Therefore, one key aspect of germline sexual identity is to regulate how GSCs respond to signals in their niche environment (Bhaskar, 2022).
Different findings have led to different conclusions about the role of the JAK/STAT pathway in male GSCs. When STAT activity is removed from individual GSCs, they are lost rapidly from the niche, indicating a role in GSC identity or maintenance. However, when STAT is removed from all GSCs, they exhibit defects in niche adhesion but can otherwise function as GSCs, although GSC loss is also observed. The JAK/STAT pathway has also been implicated in aging of GSCs and their niche. One interpretation of these diverse data would be that the JAK/STAT pathway is important for specific aspects of male GSC function, such as regulation of cell adhesion and the cell cycle, but it is not required for stem cell identity per se (Bhaskar, 2022).
A different role is proposed for the JAK/STAT pathway, which is to regulate GSC sexual identity. Previously it was reported that the JAK/STAT pathway is important for establishing male identity in the embryonic germline. This study shows that one defect observed in XX germ cells present in a male soma is that they exhibit reduced JAK/STAT signaling. Further, activation of the JAK/STAT pathway can partially rescue these XX germ cells, promoting a male identity and progression into spermatogenesis. Thus, it is proposed that the JAK/STAT pathway remains a key masculinizing signal for the germline throughout development and into adulthood. One possibility is that the JAK/STAT pathway regulates only GSC sex and that other roles, such as regulating a specific set of cell adhesion proteins, represent downstream consequences of altering sexual identity. Alternatively, the JAK/STAT pathway could regulate GSC sexual identity and other aspects of GSC behavior independently.
One important way in which the JAK/STAT pathway promotes a male identity in the germline is by activating the male sex determination factor Phf7. Previously, it was shown that Phf7 is important for male identity in the germline and proper spermatogenesis. Phf7 likely promotes male germline identity by acting as a chromatin 'reader' and binding to histone H3 methylated at position K4. Phf7 is also toxic to female germ cells, making the sex-specific regulation of Phf7 extremely important. This study shows that the JAK/STAT pathway is a direct regulator of Phf7 expression in both embryos and adults. STAT protein can bind to the Phf7 locus, and consensus STAT binding sites near the male-biased promoter are essential for proper male expression of Phf7 and its ability to function in spermatogenesis. Expression from the male-biased promoter is important in part because the transcript from the downstream, 'female' promoter is subject to translational repression. Thus, Phf7 represents an important link between the JAK/STAT pathway and male identity in the germline (Bhaskar, 2022).
Sxl acts as a key regulator of sex determination in both the soma and the germline, and it is necessary and sufficient to confer female identity. However, the role of Sxl in the germline has remained mysterious. In the soma, Sxl regulates sexual identity through tra and dosage compensation through msl-2, but these genes do not play a role in the germline. Instead, this study has found that a key role of Sxl in the germline is to repress the JAK/STAT pathway in female germ cells (Bhaskar, 2022).
Initially, only the male somatic gonad expresses ligands for the JAK/STAT pathway and is capable of promoting JAK/STAT activation in the germ cells. However, ligands for the JAK/STAT pathway eventually become active in the germarium of the ovary, where they are important for the function or maintenance of the somatic escort cells. Sxl acts to repress JAK/STAT response in the female germ cells and thereby prevents activation of male-promoting factors such as Phf7. Somatic cells of the ovary such as the escort cells are still able to respond to these ligands and activate the JAK/STAT pathway, even though they also express Sxl. How Sxl is able to repress the JAK/STAT response in a germline-specific manner remains unknown, although the levels of Sxl appear higher in the GSCs than in the surrounding soma. However, the fact that an activated Hop (hopTum) can partially rescue the germline in XX males indicates that Sxl is repressing the pathway at the level of Hop or above. Interestingly, RNA for the JAK/STAT receptor domeless was identified in a pull-down experiment with Sxl, suggesting this could be a relevant target for regulation (Bhaskar, 2022).
These data support a model where the JAK/STAT pathway is important for activating male identity in the germline and expression of male genes such as Phf7, while this pathway is repressed in female germ cells by Sxl. Loss of Sxl from the female germline leads to both upregulation of JAK/STAT signaling and inappropriate expression of Phf7. In addition, suppression of the JAK/STAT pathway can partially rescue loss of Sxl in the female germline. Thus, regulation of the JAK/STAT pathway is one key aspect of how Sxl promotes female germline identity. However, no ability was observed for loss of Phf7 to rescue loss of Sxl from the female germline. This is in contrast to previously published results where loss of Phf7 was shown to rescue the female germline in sans fille mutants, which also primarily affects the germline by disrupting Sxl expression. This study has now reduced Phf7 function by both RNAi and using null Phf7 mutants, in both Sxl and sans fille loss-of-function backgrounds and observed no rescue or modification of the germline defects present. It is concluded that, while regulation of Phf7 by the JAK/STAT pathway and Sxl is clearly important for proper germline sexual development, ectopic expression of Phf7 is not the only defect present in Sxl mutant female germ cells; there must be additional targets for regulation by Sxl and JAK/STAT that are disrupted in Sxl mutants. In support of this view, loss of STAT from the male germline has a more severe phenotype than loss of Phf7. Previously, it has been shown that expression of another male-promoting factor in the germline, Tdrd5l, is regulated by Sxl. While this regulation appears to be, at least in part, via Sxl acting on the Tdrd5l mRNA to influence levels of Tdrd5l protein, it is possible that Tdrd5l is also regulated at the transcriptional level as an additional target of the JAK/STAT pathway (Bhaskar, 2022).
It is intriguing that Sxl acts as a negative regulator of the JAK/STAT pathway in both the soma and the germline but does so in different ways. In the soma, Sxl activates an alternative splicing cascade that leads to splicing of dsx in the female mode, creating the DSXF protein, while the DSXM protein is produced in males by default. An important sex-specific trait in the embryonic gonad is that male somatic cells produce ligands for the JAK/STAT pathway that activate JAK/STAT signaling specifically in male germ cells, and this is regulated in a manner dependent on dsx. Thus, in addition to being a negative regulator of JAK/STAT signal reception in the germline, Sxl acts as a negative regulator of JAK/STAT ligand production in the soma. Together, these independent aspects of regulation by Sxl combine to ensure that the masculinizing effects of the JAK/STAT pathway are restricted to male germ cells (Bhaskar, 2022).
An important conclusion from this work is that germline sex determination regulates how GSCs communicate with their surrounding stem cell niche. Germline sex determination is regulated by both germline autonomous cues, based on the germline sex chromosome constitution, and non-autonomous signals from the soma. The autonomous cues, acting through Sxl, regulate how signals from the niche are received and interpreted by the GSCs. In both the testis and ovary GSC cell niches, the JAK/STAT pathway is important for regulating somatic cells like the cyst stem cells in the testis and the escort cells in the ovary. However, this pathway is only required in the male GSCs and not female GSCs. It is proposed that it is essential to block JAK/STAT signaling in female GSCs to prevent their exposure to this masculinizing signal. Indeed, activation of the JAK/STAT signal is sufficient to promote male identity in XX germ cells, and removal of STAT is sufficient to partially rescue the defects observed in XX germ cells that have lost Sxl. Thus, a key aspect of how Sxl promotes female identity in the germline is to prevent female GSCs from being masculinized by activators of the JAK/STAT pathway present in the niche environment (Bhaskar, 2022).
It is important to note that, when the sex chromosome genotype affecting germline is referred to as 'sex determination' this could result from any contribution of sex chromosome genotype to successful spermatogenesis or oogenesis. For example, if dosage compensation is incomplete or non-existent in the germline, then the presence of two X chromosomes will lead to increased X chromosome gene expression, which may be incompatible with male germline differentiation. Similarly, a single X chromosome dose may be incompatible with oogenesis. It is also possible that the number of X chromosomes present in the germline has additional affects besides the presence or absence of Sxl expression. While XX germ cells present in a male soma exhibit severe atrophy and loss, the expression of Sxl in the male germline has a much weaker phenotype. Thus, there may be additional consequences of sex chromosome genotype on germline function beyond that which is controlled by Sxl. A better understanding of what germline sexual identity means in Drosophila, in particular at the level of whole-genome gene expression levels, is required before it will be possible to assess the true contribution of germline sex chromosome constitution to germline sex determination. Further, how the effects of X chromosome number on germline sexual development in Drosophila relate to infertility observed in patients with disorders of sexual development such as Klinefelter's and Turner's syndromes remains to be investigated (Bhaskar, 2022).
One limitation of this study is the relatively low frequency with which it was possible to rescue the XX germline in tra mutants by downregulating Sxl or upregulating the JAK/STAT pathway. This could be due to technical limitations of the timing or level of expression of the reagents used. Alternatively, this could mean that there is another interesting defect in XX germ cells present in a male somatic environment besides that is caused by expression of Sxl and downregulation of the JAK/STAT pathway. Another limitation of the study is that the molecular target for Sxl in regulating the JAK/STAT pathway remains unknown. RNA-seq analysis of Bam mutant testes and ovaries suggested that hop RNA splicing is differentially regulated between males and females. However, extensive experimental analysis failed to reveal a role for Sxl in regulating hop in the germline. Thus, this remains an important area for future research (Bhaskar, 2022).
N6-methyladenosine (m6A) is the most common internal modification of eukaryotic messenger RNA (mRNA) and is decoded by YTH domain proteins. Drosophila mRNA m6A methylosome consists of Ime4 and KAR4 (Inducer of meiosis 4 and Karyogamy protein 4), and Female-lethal (2)d (Fl(2)d) and Virilizer (Vir). In Drosophila, fl(2)d and vir are required for sex-dependent regulation of alternative splicing of the sex determination factor Sex lethal (Sxl). However, the functions of m6A in introns in the regulation of alternative splicing remain uncertain. This study shows that m6A is absent in the mRNA of Drosophila lacking Ime4. In contrast to mouse and plant knockout models, Drosophila Ime4-null mutants remain viable, though flightless, and show a sex bias towards maleness. This is because m6A is required for female-specific alternative splicing of Sxl, which determines female physiognomy, but also translationally represses male-specific lethal 2 (msl-2) to prevent dosage compensation in females. The m6A reader protein YT521-B decodes m6A in the sex-specifically spliced intron of Sxl, as its absence phenocopies Ime4 mutants. Loss of m6A also affects alternative splicing of additional genes, predominantly in the 5' untranslated region, and has global effects on the expression of metabolic genes. The requirement of m6A and its reader YT521-B for female-specific Sxl alternative splicing reveals that this hitherto enigmatic mRNA modification constitutes an ancient and specific mechanism to adjust levels of gene expression (Haussmann, 2016).
N6-methyladenosine RNA (m6A) is a prevalent messenger RNA modification in vertebrates. Although its functions in the regulation of post-transcriptional gene expression are beginning to be unveiled, the precise roles of m6A during development of complex organisms remain unclear. This study carried out a comprehensive molecular and physiological characterization of the individual components of the methyltransferase complex, as well as of the YTH domain-containing nuclear reader protein in Drosophila melanogaster. The member of the split ends protein family, Spenito, was identified as a novel bona fide subunit of the methyltransferase complex. Important roles of this complex were demonstrated in neuronal functions and sex determination, and the nuclear YT521-B protein was implicated as a main m6A effector in these processes. Altogether, this work substantially extends knowledge of m6A biology, demonstrating the crucial functions of this modification in fundamental processes within the context of the whole animal (Lence, 2016).
Components of the methyltransferase complex have been shown to be essential during early development of various organisms. In contrast to these studies, the current analysis argues against a vital role for Ime4 in Drosophila as both deletion alleles give rise to homozygous adults without prominent lethality during development. This cannot be explained by compensation via dMettl14, as its knockout produces similar effects as the Ime4 (Methyltransferase like 3) knockout. Furthermore, depleting both genes only slightly intensifies Mthe locomotion phenotype without affecting fly survival, supporting the idea that Ime4 and dMettl14 act together to regulate the same target genes. Accordingly, loss of either component in vivo dramatically affects stability of the other (Lence, 2016).
Loss of function of either of the methyltransferases produces severe behavioural defects. All of them can be rescued by specific expression of Ime4 cDNA in the nervous system of Ime4 mutants, indicating neuronal functions. This is consistent with the substantial enrichment of m6A and its writer proteins in the embryonic neuroectoderm, as well as with the affected genes upon depletion in S2R+ cells. These analyses further reveal notable changes in the architecture of NMJs, potentially explaining the locomotion phenotype. In the mouse, m6A is enriched in the adult brain, whereas in zebrafish, METTL3 and WTAP show high expression in the brain region of the developing embryo. Furthermore, a crucial role for the mouse m6A demethylase FTO in the regulation of the dopaminergic pathway was clearly demonstrated. Thus, together with previous studies, this work reveals that m6A RNA methylation is a conserved mechanism of neuronal mRNA regulation contributing to brain function (Lence, 2016).
This study found that Ime4 and dMettl14 also control the splicing of the Sxl transcript, encoding for the master regulator of sex determination in Drosophila. This is in agreement with the previously demonstrated roles of Fl(2)d and Vir in this process. However, in contrast to these mutants, mutants for Ime4, dMettl14 and YT521-B are mostly viable, ruling out an essential role in sex determination and dosage compensation. Only when one copy of Sxl is removed, Ime4 mutant females start to die. Notably, m6A effect on Sxl appears more important in the brain compared to the rest of the organism, possibly allowing fly survival in the absence of this modification (Lence, 2016).
A targeted screen identifies Nito as a bona fide methlytransferase complex subunit. The vertebrate homologue of Spenito, RBM15, was recently shown to affect XIST gene silencing via recruitment of the methyltransferase complex to XIST RNA, indicating that its role in m6A function and dosage compensation is conserved40. In summary, this study provides a comprehensive in vivo characterization of m6A biogenesis and function in Drosophila, demonstrating the crucial importance of the methyltransferase complex in controlling neuronal functions and fine-tuning sex determination via its nuclear reader YT521-B (Lence, 2016).
Sexual dimorphisms in body size are widespread throughout the animal
kingdom but their underlying mechanisms are not well characterized. Most
models for how sex chromosome genes specify size dimorphism have
emphasized the importance of gonadal hormones and cell-autonomous
influences in mammals versus strictly cell-autonomous mechanisms in
Drosophila melanogaster. This study used tissue-specific genetics to
investigate how sexual size dimorphism (SSD) is established in
Drosophila. The larger body size characteristic of Drosophila females is
established very early in larval development via an increase in the
growth rate per unit of body mass. The female sex determination gene Sex-lethal (Sxl) was shown to function in
central nervous system (CNS) neurons as part of a relay that
specifies the early sex-specific growth trajectories of larval but not
imaginal tissues. Neuronal Sxl acts additively in 2 neuronal
subpopulations, one of which corresponds to 7 median neurosecretory
cells: the insulin-producing cells (IPCs). Surprisingly, however,
male-female differences in the production of insulin-like peptides
(Ilps) from the IPCs do not appear to be involved in establishing SSD in
early larvae, although they may play a later role. These findings
support a relay model in which Sxl in neurons and Sxl in local tissues
act together to specify the female-specific growth of the larval body.
They also reveal that, even though the sex determination pathways in
Drosophila and mammals are different, they both modulate body growth via
a combination of tissue-autonomous and nonautonomous inputs (Sawala, 2017).
The sex of an organism has profound effects on its morphology, physiology, and behaviour. It also influences the risk of developing diseases of growth and metabolism such as obesity, metabolic syndrome, cardiovascular disease, and cancer. One important feature of sexual dimorphism is the difference in body size between males and females, called sexual size dimorphism (SSD). SSD is widespread and rapidly evolving across the animal kingdom such that, depending upon the species, the larger sex can either be male or female. It is not yet clear, however, which mechanisms link the chromosomal sex of an organism to specific male and female patterns of growth during development and thus ultimately to adult SSD (Sawala, 2017).
In mammals, the presence or absence of a single gene on the male Y chromosome (Sry) determines gonad differentiation into male testis or female ovary, respectively. The gonads then establish a sex-specific hormonal milieu that controls male and female differentiation, patterns of growth, and metabolism. However, recent studies in mice now demonstrate that the cellular (i.e., chromosomal) sex of nongonadal tissues can also influence growth and metabolism, an effect that is thought to be modified by the action of gonadal hormones. Hence, although many details remain to be explored, it is likely in mammals that patterns of growth relevant to SSD are controlled via both hormonal and cell-autonomous mechanisms (Sawala, 2017).
The fruit fly Drosophila melanogaster has provided insights into many aspects of sexual dimorphism. In Drosophila, sex determination is based on X chromosome dosage, with XX individuals developing as females and XY individuals as males. At the early embryonic stage, the presence of 2 X chromosomes in females activates the expression of sex determination gene Sex-lethal (Sxl), which is thereafter maintained via positive autoregulation. Sxl controls the splicing of multiple downstream targets regulating sexual differentiation as well as X chromosome dosage compensation. A commonly held view is that Drosophila, unlike mammals, deploys sex determination genes in a strictly cell-autonomous manner such that they are required in every somatic cell that is sexually dimorphic. Nevertheless, there are hints that non-cell-autonomous mechanisms regulate at least some aspects of sexual dimorphism. For example, in adult flies, ecdysone and juvenile hormone can act like sex hormones to regulate reproduction and sexual identity (Sawala, 2017).
SSD in Drosophila is present at larval, pupal, and adult stages, with females approximately 30% larger than males. Drosophila larvae are composed of 2 types of tissues: polyploid larval-specific organs that are histolysed during pupation and diploid imaginal tissues, which are the precursors of adult structures. Polyploid tissues comprise the bulk of the larva such that body SSD measured at larval stages reflects their growth rather than that of imaginal tissues. In contrast, body SSD measured at the adult stage reflects the larval/pupal growth of imaginal tissues. SSD appears to be present in most tissues and may be a result of differences in both cell size and number. Signalling pathways important for body growth, such as the insulin/insulin-like growth factor (IGF) and target of rapamycin (Tor) network, are likely to be relevant for SSD but precisely how sex modulates them remains unclear. In terms of sex determination genes, SSD was long thought to be dependent upon Sxl but not on one of its key downstream target genes, transformer (tra), which specifies other aspects of sexual dimorphism. This was challenged recently by a report that tra contributes to SSD, although there is also likely to be a tra-independent mechanism. The same study also reported that tra not only exerts autonomous effects on cell size but also acts in the female fat body to stimulate the secretion of insulin-like peptides (Ilps) from the brain, which in turn promotes larger body size in females (Sawala, 2017).
This study investigated the link between sex determination genes and SSD in Drosophila using cell type-specific genetic manipulations and size measurements of both larval and imaginal tissues. Surprisingly, it was found that Sxl controls SSD by acting in specific subsets of female neurons to increase body growth during larval development. At this stage, neuronal Sxl functions selectively to increase the growth rate of larval but not imaginal tissues. This study reveals that the sex of specific neurons regulates SSD in a non-cell-autonomous manner (Sawala, 2017).
Sex-specific body sizes are established after embryogenesis but early during larval development. SSD is manifested as a higher growth rate in female than male larvae, with a maximal difference occurring during L2 and early L3. Previous SSD studies focused later in development, after critical weight (CW), a key checkpoint in L3 that can influence final body size. The study by Testa (2013) showed that females are larger as they attain a higher CW and then a higher absolute growth rate during subsequent larval development (the terminal growth period). The data are consistent with this but also reveal that the establishment of different growth patterns in females and males involves an early divergence of mass-specific growth rates (mass gain per h per mg of body mass) in L2, well before CW. After SSD has been established in L2/early L3, it is subsequently maintained as larger female larvae gain more absolute mass per hour than smaller males, although both sexes now have very similar mass-specific growth rates (Sawala, 2017).
The insulin signalling pathway is a major stimulator of larval growth in Drosophila and it has been reported that IPC secretion of Ilp2 and InR signalling are both higher in females than in males at the L3 stage (Testa, 2013). However, at earlier stages relevant for the establishment of SSD, this study did not detect any male-female differences in Ilp2 secretion or in insulin signalling. Furthermore, larvae with decreased Ilp production from IPCs or larvae lacking all 3 of the Ilp genes normally expressed in the IPCs are smaller but still retain larval body SSD. Thus, insulin signalling is required for maximal larval growth in both males and females but it does not appear to be a sex-specific regulator of larval SSD. Interestingly, however, SSD of the developing wing disc is decreased by InR knockdown or decreased insulin production from IPCs. Thus, although insulin signalling may not contribute to the SSD of larval tissues, it does regulate that of imaginal tissues. Higher insulin signalling in female imaginal tissues could be driven by local Sxl/tra dependent modulation of insulin sensitivity and perhaps from mid/late L3 stages by increased Ilp production. Very recently, it was reported that another key growth regulator, Myc, is more highly expressed in female than in male larvae and so may contribute to SSD (Wehr Mathews, 2017). Sex differences in myc transcript expression may be the result of this X-linked gene escaping complete dosage compensation rather than via direct Sxl regulation. Interestingly, this study found that the higher expression of myc characteristic of females is fully maintained in elavc155>Sxl RNAi larvae, even though they are masculinised in terms of larval growth. This indicates that a sex difference in global myc levels is not sufficient to confer SSD. It remains possible, however, that myc in neurons or in another tissue has a role in SSD, potentially downstream of neuronal Sxl (Sawala, 2017).
A central finding of this study is that Sxl is required in the CNS to direct the female-specific growth trajectory of the larval body. By mapping the site of action of Sxl in the CNS using a panel of Gal4 drivers, this study was able to show that it functions additively in 2 nonoverlapping populations of neurons within the CNS: Gad1-Gal4 neurons and IPCs. For the Gad1-Gal4 neurons, it remains to be determined whether GABA+ or GABA- subsets are relevant for SSD and whether or not they functionally interconnect with IPCs in the larval CNS. Nevertheless, it has been reported for the adult CNS that some GABAergic neurons converge on IPCs, which express the metabotropic GABAB receptor and may respond to GABA by decreasing Ilp secretion. Importantly, this study also mapped another critical site of action for Sxl to the IPCs, a cluster of only 7 peptidergic neurons. This finding, together with the evidence that sex-specific IPC secretion of Ilps is unlikely to establish SSD, suggests that one or more of the numerous other neuropeptides/secreted factors expressed in larval IPCs may be relevant (Sawala, 2017).
This study found that neuronal Sxl appears to regulate SSD largely independently of its best-characterised downstream targets, tra and msl-2. The lack of a major neuronal tra input in SSD makes it unlikely that there is a role for its neuronal target fruitless, which regulates many aspects of sex-specific behaviour in adults. In addition, sex-specific isoforms of Fruitless have not been detected until the late L3 stage, long after SSD has been established. New direct Sxl targets have been identified in recent years in several biological contexts and future approaches targeting the early larval nervous system may reveal how neuronal Sxl controls SSD. Importantly, whatever the Sxl targets in neurons relevant for SSD turn out to be, the current results clearly demonstrate that Sxl acts in a remote manner to regulate peripheral tissue growth and overall body size. Identification of the complete remote-control pathway is beyond the scope of this study but, in principle, it could involve the neural secretion of a hormone-like signal and/or innervation of peripheral organ(s) that regulate growth. (Sawala, 2017).
A previous report implicated the Sxl target tra acting in the fat body as an important non-cell-autonomous regulator of female body size (Rideout, 2015). The current study, however, finds that the genetic requirement for Sxl in body size is stronger in neurons (likely acting without tra) than it is in the fat body (acting with tra). Not all of the conclusions made in the two studies are easily reconcilable, but it is noted that the previous report used body size readouts that were adult mass or estimated pupal volume, whereas this study measured larval body mass during L2 and L3. It nevertheless remains possible that Sxl can act in both neurons and fat body to regulate overall body SSD in a non-tissue-autonomous manner. Perhaps the relative contributions of each site of Sxl expression depend upon diet or other factors that varied between the 2 studies. Either way, the current finding that restoration of Sxl expression specifically in neurons is sufficient to increase the body size of Sxl mutant females demonstrates that the neuronal relay mechanism is functional without Sxl in the fat body (Rideout, 2017).
Evidence supporting a neuronal relay model for SSD in Drosophila is discussed. Central to this model is the finding that Sxl in neurons is required to relay a signal for female body size to peripheral larval tissues. Thus, loss of Sxl in neurons abolishes SSD by decreasing the larval body mass of females to that of males. Conversely, neuronal Sxl expression is sufficient for substantial rescue of female body size in Sxl mutant larvae. Neuronal Sxl knockdown decreases SSD in larvae at the level of individual polyploid larval tissues such as the fat body, but, surprisingly, this does not appear to be the case for the diploid wing imaginal disc. Nevertheless, after completion of pupal development, neuronal Sxl depletion does eventually lead to decreased SSD of the adult wings as well as the adult body. So, how is it possible to account for why the neuronal Sxl input affects imaginal tissue SSD at adult but not larval stages? One possible explanation is that neuronal Sxl functions during pupal stages to nonautonomously regulate imaginal tissue growth. Alternative explanations involve neuronal Sxl acting during larval stages to specify pupal resources/signals that themselves regulate the SSD of adult body structures. For example, nutrient resources laid down in larval tissues can be mobilized by the process of histolysis during the pupal period. Hence, the greater mass of the female larva may be needed to sustain the greater final size of the female adult. An important feature of this relay model for SSD is that the Sxl-dependant signal from neurons is integrated with local Sxl/tra inputs in both larval and imaginal tissues. Evidence for these tissue-autonomous Sxl/tra growth inputs comes from previous studies as well as the current study finding that Sxl and tra activities in the larval fat body and wing imaginal disc contribute towards the increased size characteristic of these female tissues. It was also found that Sxl and Tra were unable to increase fat body cell size in a cell-autonomous manner significantly in males or in females lacking neuronal Sxl. Hence, larval tissue-autonomous Sxl/TraF activity may only be able to boost growth efficiently in the presence of the Sxl-dependent signal from neurons. It is therefore proposed that Sxl activity in neurons and in local tissues acts together to maximize female tissue growth (Rideout, 2017).
A widely held textbook view is that somatic sexual dimorphism is regulated very differently in Drosophila and mammals: the former in a cell-autonomous manner, the latter by gonad-derived hormones. However, the discovery that the brain remotely regulates SSD in Drosophila may have its counterpart in mammals. Thus, mammalian gonadal sex steroids are thought to act on neurons in the hypothalamus to regulate growth hormone secretion from the anterior pituitary gland. Male-female differences in the levels and patterns of circulating growth hormone are thought to induce sex-specific insulin-like growth factor 1 (IGF-1) profiles, in turn conferring dimorphic growth patterns. Growth hormone itself does not appear to be conserved in Drosophila, but it is interesting that the pars intercerebralis of the Drosophila brain, harbouring the IPCs, has been likened to the mammalian hypothalamus. Furthermore, IPCs send projections to the ring gland, which has been likened to the pituitary gland in mammals. In a second parallel, the current findings together with several studies in mammals suggest that primary sex determination signals act via a combination of cell-autonomous and non-cell-autonomous mechanisms to control overall SSD. For example, in human and other mammalian embryos, sex differences in body size are noticeable before gonad differentiation, suggesting the existence of gonadal sex hormone-independent mechanisms of SSD. Moreover, an XX rather than an XY chromosome complement in mice can increase adult body mass by approximately 7%, independently of gonadal sex, and this difference can be strongly enhanced by gonadectomy. In conclusion, the upstream sex determination pathways in Drosophila and mammals are very different but the regulatory logic of how they regulate body growth via both cell-autonomous and remote mechanisms appears to be more similar than previously thought (Rideout, 2017).
Somatic sexual determination and behavior in Drosophila melanogaster are under the control of a genetic cascade initiated by Sex lethal (Sxl). In the female soma, SXL RNA binding-protein regulates the splicing of transformer (tra) transcripts into a female-specific form. The RNA binding protein TRA and its cofactor TRA2 function in concert in females, whereas SXL, TRA and TRA2 are thought not to function in males. To better understand sex-specific regulation of gene expression, this study analyzed male and female head transcriptome datasets for expression levels and splicing, quantifying sex-biased gene expression via RNA-Seq and qPCR. The data uncouples the effects of Sxl and tra/tra2 in females in the sex-biased alternative splicing of head transcripts from the X-linked locus found in neurons (fne), encoding a pan-neuronal RNA-binding protein of the ELAV family. FNE protein levels are down regulated by Sxl in female heads, also independently of tra/tra2. It is argued that this regulation may have important sexually dimorphic consequences for the regulation of nervous system development or function (Sun, 2015).
The Drosophila sex determination hierarchy is the classical model of developmentally regulated alternative splicing. To identify genes expressed differentially in males and females, head samples were chosen, thereby eliminating large numbers of events restricted to gonadal differentiation. Moreover, the neurons, enriched in heads, are the site of extensive regulation at the level of alternative splicing (Sun, 2015).
In addition to dsx and fru, canonical regulators of Drosophila sex determination, we identified and further characterized the expression of fne and tango13 as genes expressed in a sex-biased manner. This study found that tango13 sex-specific expression responds to tra, tra2, and Sxl mutations in females as expected if under the control of the canonical sex determination pathway. An intriguing feature is the absence of reciprocity in the regulation of the mutually exclusive tango13-a and tango13-b splice forms, because tango13-a levels are reduced in tra, tra2, and Sxl mutants, but tango13-b levels are not. This observation suggests that tra and tra2 could possibly have an impact on the levels/stability of the tango13-a transcript rather than on the alternative splicing of tango13 RNA per se. The impact of tra/tra2 alleles on the expression of tango13-a is similar to that on the expression of dsx-F, consistent with regulation downstream of the sex determination pathway (Sun, 2015).
In contrast to fru, dsx, and tango13, the expression of fne is independent of TRA and TRA2. Crucially, fne splicing nevertheless depends on Sxl function in female heads: Sxl- pseudomales switch to a male mode of fne alternative splicing, consistent with a role for SXL in promoting, directly or indirectly, the formation of the fne-b isoform at the expense of fne-a in normal females. Further, although fne alternative splicing is male-like in XX Sxl pseudomales, FNE protein levels are also upregulated two-fold to three-fold compared to CS males and females. Both male-like splicing and increased FNE protein levels in the pseudomales are reverted by the introduction of a Sxl+ minigene, confirming the specificity of Sxl in the control of both the splicing and protein levels. These data thus show that a Sxl-dependent, tra/tra2-independent mechanism regulates fne expression in females (Sun, 2015).
Canton-S (CS) female and male pools of fne RNA yield similar amounts of FNE protein in the two sexes. However, XX SxlM1,fΔ33 / Sxlf7M1 pseudomales have a male-like pool of fne RNA and two-fold to three-fold increased FNE protein levels compared to CS. Because fne is an X-linked gene, its expression is presumably influenced by the canonical dosage compensation pathway, which could be responsible for the upregulation of FNE levels in XX Sxl- pseudomales. However, according to the canonical model, higher fne transcript levels would be expected in pseudomales than in males and females, but that is not the case. Additional mechanisms must be at play (Sun, 2015).
First, increased FNE protein levels in XX Sxl- pseudomales compared to wild-type males do not result from increased transcript levels. Because males and pseudomales share similar spliced pools of fne RNA, their distinct FNE outputs necessarily result from a regulatory mechanism that operates independently of the effects of Sxl on alternative splicing. Formally, this mechanism appears to stimulate the translation of fne transcripts in XX individuals. Second, increased FNE protein levels, concomitant with changes in alternative splicing but not associated with changes in transcript levels, as in XX Sxl- pseudomales compared to wild-type females, are consistent with the existence of a Sxl-dependent mechanism that downregulates FNE protein levels in XX females. Only the XX-dependent upregulation would persist in Sxl- pseudomales, hence their increased FNE level. It is conceivable that fne regulation by Sxl occurs via direct binding of Sxl to fne transcripts. An interesting alternative as a means to regulate its splicing is the possibility that the impact of Sxl on fne expression occurs indirectly (possibly via an hormonal axis), since the extensive impact of the germline on the expression of somatic genes has been documented (Sun, 2015).
fne encodes an RNA-binding protein concentrated in the soma of neurons and present throughout development. It is necessary for the normal development of the mushroom bodies of males and females, and it is involved in the regulation of male courtship. It is intriguing that the expression of pan-neuronal fne is regulated in a sex-biased manner under the control of Sxl (Sun, 2015).
In addition to its role in the development of the germline, Sxl is involved in several regulatory pathways in the soma. It responds to a cell autonomous signal (number of X chromosomes) and is crucial both for the sexual development of somatic cells and for dosage compensation in males. SXL, but not TRA or TRA2, is also required independently of the somatic sex determination pathway for the development of a subset of sexually dimorphic neurons, with consequences on female ovulation. Additional phenotypes independent of the canonical somatic sex determination pathway but dependent on Sxl are the control of the sexually dimorphic body size of flies and the sex-specific bristle number on the A5 sternite. The latter occurs through general downregulation of the Notch pathway by SXL in multiple tissues . Thus, the Sxl-regulated expression of fne fits within the context of Sxl acting in parallel with the canonical Sxl-tra/tra2 cascade, constituting an example of its impact on tissues that do not show obvious sexual dimorphism (Sun, 2015).
fne is a member of a fairly new multigene family restricted to dipterans. The birth of this family predates the role of SXL in sex determination, which is restricted to the drosophilids. Based on RNA-Seq data and the Flybase models, sex-specific alternative splicing has not been reported for either of the other two paralogues in this family, elav (embryonic lethal abnormal visual system, X linked) or rbp9 (RNA binding protein 9, second chromosome). elav is the result of a retrotransposition and is likely to have acquired new cis-regulatory elements in the process. It autoregulates via a posttranscriptional mechanism involving its 3' UTR. It is unclear whether the Sxl-dependent regulation of fne is an ancestral property that has been lost for rbp9 or was recently acquired. Nevertheless, sex-specific alternative splicing provides fne with the ability to be differentially regulated in females, which may have an important impact on sex-specific nervous system function or development, for which there are numerous instances of a role for Sxl. Within the context of the canonical sex determination pathway, Sxl regulates the expression of fru and dsx, two transcription factors crucial for behavior and nervous system function. SXL also controls, via an independent pathway, specific aspects of female behavior (Evans and Cline 2013). Still outside of the context of the canonical sex determination pathway, Sxl regulates the neurogenic locus Notch. Further, in Drosophila virilis, SXL protein accumulates in the male developing nervous system, consistent with a role there . Thus, the control exerted by Sxl on pan-neuronal fne outside of the context of the canonical sex determination pathway may be part of the heritage of an SXL ancestral function more focused on the nervous system than on sexual differentiation (Sun, 2015).
Reproduction in sexually dimorphic animals relies on successful gamete production, executed by the germline and aided by somatic support cells. Somatic sex identity in Drosophila is instructed by sex-specific isoforms of the DMRT1 ortholog Doublesex (Dsx). Female-specific expression of Sex-lethal (Sxl) causes alternative splicing of transformer (tra) to the female isoform traF. In turn, TraF alternatively splices dsx to the female isoform dsxF. Loss of the transcriptional repressor Chinmo in male somatic stem cells (CySCs; cyst stem cells) of the testis causes them to "feminize", resembling female somatic stem cells in the ovary. This somatic sex transformation causes a collapse of germline differentiation and male infertility. This feminization occurs by transcriptional and post-transcriptional regulation of traF. chinmo-deficient CySCs upregulate tra mRNA as well as transcripts encoding tra-splice factors Virilizer (Vir) and Female lethal (2)d (Fl(2)d). traF splicing in chinmo-deficient CySCs leads to the production of DsxF at the expense of the male isoform DsxM, and both TraF and DsxF are required for CySC sex transformation. Surprisingly, CySC feminization upon loss of chinmo does not require Sxl but does require Vir and Fl(2)d. Consistent with this, this study shows that both Vir and Fl(2)d are required for tra alternative splicing in the female somatic gonad. This work reveals the need for transcriptional regulation of tra in adult male stem cells and highlights a previously unobserved Sxl-independent mechanism of traF production in vivo. In sum, transcriptional control of the sex determination hierarchy by Chinmo is critical for sex maintenance in sexually dimorphic tissues and is vital in the preservation of fertility (Brmai, 2018).
This study shows that that one single factor, Chinmo, preserves the male identity of adult CySCs in the Drosophila testis by regulating the levels of canonical sex determinants. CySCs lacking chinmo lose DsxM expression not by transcriptional loss but rather by alternative splicing of dsx pre-mRNA into dsxF. These chinmo-mutant CySCs ectopically express TraF and DsxF, and both factors are required for their feminization. Furthermore, the results demonstrate that tra alternative splicing in cyst cells lacking chinmo is achieved independently of Sxl. Instead, this work strongly suggests that traF production in the absence of chinmo is mediated by splicing factors Vir and Fl(2)d. It is proposed that male sex identity in CySCs is maintained by a two-step mechanism whereby traF is negatively regulated at both transcriptional and post-transcriptional levels by Chinmo (see Model for adult somatic sex maintenance in the Drosophila somatic gonad). In this model, loss of chinmo from male somatic stem cells first leads to transcriptional upregulation of tra pre-mRNA as well as of vir and fl(2)d. Then the tra pre-mRNA in these cells is spliced into traF by the ectopic Vir and Fl(2)d proteins. The ectopic TraF in chinmo-deficient CySCs then splices the dsx pre-mRNA into dsxF, resulting in loss of DsxM and gain of DsxF, and finally induction of target genes usually restricted to follicle cells in the ovary (Brmai, 2018).
Chinmo has motifs associated with transcriptional repression and its loss clonally is associated with ectopic transcription. One interpretation of the data is that Chinmo directly represses tra, vir, and fl(2)d in male somatic gonadal cells. As the binding site and potential co-factors of Chinmo are not known, future work will be needed to determine whether Chinmo directly regulates expression of these genes. It is also noted that ~50% of chinmo-mutant testes still feminize in the genetic absence of tra or dsxF. These latter data indicate that Chinmo regulates male sex identity through another, presumably parallel, mechanism that does not involve canonical sex determinants. However, this tra/dsx-independent mode of sex maintenance downstream of Chinmo is not characterized and will require the identification of direct Chinmo target genes (Brmai, 2018).
Previous work has shown that JAK/STAT signaling promotes chinmo in several cell types, including CySCs (Flaherty, 2010). Since JAK/STAT signaling is itself sex-biased and restricted to the embryonic male gonad, it is presumed that activated Stat92E establishes chinmo in male somatic gonadal precursors, perhaps as early as they are specified in the embryo. Because loss of Stat92E from CySCs does not result in an apparent sex transformation phenotype, the interpretation is favored that Stat92E induces expression of chinmo in CySCs but that other sexually biased factors maintain it. One potential candidate is DsxM, which is expressed specifically in early somatic gonads and at the same time when Stat92E activation is occurring in these cells. In fact, multiple DsxM ChIP-seq peaks were identified in the chinmo locus, suggesting potential regulation of chinmo by DsxM. This suggests a potential autoregulatory feedback loop whereby DsxM preserves its own expression in adult CySCs by maintaining Chinmo expression, which in turn prevents traF and dsxF production (Brmai, 2018).
Recent studies on tissue-specific sex maintenance demonstrate that while the Sxl/Tra/Dsx hierarchy is an obligate and linear circuit during embryonic development, at later stages it is more modular than previously appreciated. For example, Sxl can regulate female-biased genes in a tra-independent manner. Additionally, Sxl and TraF regulate body size and gut plasticity independently of the only known TraF targets, dsx and fru. Negative regulation of the TraF-DsxF arm of this cascade is required to preserve male sexual identity in CySCs but unexpectedly is independent of Sxl. Because depletion of Vir or Fl(2)d significantly blocks sex transformation and both are required for tra alternative splicing in the ovary, this work reveals they can alternatively splice tra pre-mRNA even in the absence of Sxl. This is the first demonstration of Sxl-independent, Tra-dependent feminization. These results raise the broader question of whether other male somatic cells have to safeguard against this novel mechanism. Because recent work has determined that sex maintenance is important in systemic functions regulated by adipose tissue and intestinal stem cells, it will be important to determine whether Chinmo represses traF in these settings. Finally, since the transcriptional output of the sex determination pathway is conserved from Drosophila (Dsx) to mammals (DMRT1), it is possible that transcriptional regulation of sex determinants plays a similar role in adult tissue homeostasis and fertility in higher organisms (Brmai, 2018).
Eukaryotic chromosomes are spatially segregated into topologically associating domains (TADs). Some TADs are attached to the nuclear lamina (NL) through lamina-associated domains (LADs). This study identified LADs and TADs at two stages of Drosophila spermatogenesis - in bamΔ86 mutant testes which is the commonly used model of spermatogonia (SpG) and in larval testes mainly filled with spermatocytes (SpCs). This study found that initiation of SpC-specific transcription correlates with promoters' detachment from the NL and with local spatial insulation of adjacent regions. However, this insulation does not result in the partitioning of inactive TADs into sub-TADs. It was also revealed an increased contact frequency between SpC-specific genes in SpCs implying their de novo gathering into transcription factories. In addition, the specific X chromosome organization was uncovered in the male germline. In SpG and SpCs, a single X chromosome is stronger associated with the NL than autosomes. Nevertheless, active chromatin regions in the X chromosome interact with each other more frequently than in autosomes. Moreover, despite the absence of dosage compensation complex in the male germline, randomly inserted SpG-specific reporter is expressed higher in the X chromosome than in autosomes, thus evidencing that non-canonical dosage compensation operates in SpG (Ilyin, 2022).
Male killing is a selfish reproductive manipulation caused by symbiotic bacteria, where male offspring of infected hosts are selectively killed. The underlying mechanisms and the process of their evolution are of great interest not only in terms of fundamental biology, but also their potential applications. The two bacterial Drosophila symbionts, Wolbachia and Spiroplasma, have independently evolved male-killing ability. This raises the question whether the underlying mechanisms share some similarities or are specific to each bacterial species. This study analysed pathogenic phenotypes of D. bifasciata infected with its natural male-killing Wolbachia strain and compare them with those of D. melanogaster infected with male-killing Spiroplasma. Male progeny infected with the Wolbachia strain died during embryogenesis with abnormal apoptosis. Interestingly, male-killing Wolbachia infection induces DNA damage and segregation defects in the dosage-compensated chromosome in male embryos, which are reminiscent of the phenotypes caused by male-killing Spiroplasma in D. melanogaster. By contrast, host neural development seems to proceed normally unlike male-killing Spiroplasma infection. These results demonstrate that the dosage-compensated chromosome is a common target of two distinct male killers, yet Spiroplasma uniquely evolved the ability to damage neural tissue of male embryos (Harumoto, 2018a).
Several lineages of symbiotic bacteria in insects selfishly manipulate host reproduction to spread in a population, often by distorting host sex ratios. Spiroplasma poulsonii is a helical and motile, Gram-positive symbiotic bacterium that resides in a wide range of Drosophila species. A notable feature of S. poulsonii is male killing, whereby the sons of infected female hosts are selectively killed during development. Although male killing caused by S. poulsonii has been studied since the 1950s, its underlying mechanism is unknown. This study identified an S. poulsonii protein, designated Spaid, whose expression induces male killing. Overexpression of Spaid in D. melanogaster kills males but not females, and induces massive apoptosis and neural defects, recapitulating the pathology observed in S. poulsonii-infected male embryos. The data suggest that Spaid targets the dosage compensation machinery on the male X chromosome to mediate its effects. Spaid contains ankyrin repeats and a deubiquitinase domain, which are required for its subcellular localization and activity. Moreover, a laboratory mutant strain of S. poulsonii was found with reduced male-killing ability and a large deletion in the spaid locus. This study has uncovered a bacterial protein that affects host cellular machinery in a sex-specific way, which is likely to be the long-searched-for factor responsible for S. poulsonii-induced male killing (Harumoto, 2018b).
Sex-lethal (Sxl) encodes the master regulator of the sex determination pathway in Drosophila and acts by controlling sex identity in both soma and germ line. In females Sxl maintains its own expression by controlling the alternative splicing of its own mRNA. This study identifies a novel sex determination gene, spenito (nito) that encodes a SPEN family protein. Loss of nito activity results in stem cell tumors in the female germ line as well as female-to-male somatic transformations. It was shown that Nito is a ubiquitous nuclear protein that controls the alternative splicing of the Sxl mRNA by interacting with Sxl protein and pre-mRNA, suggesting that it is directly involved in Sxl auto-regulation. Given that SPEN family proteins are frequently mutated in cancers, these results suggest that these factors might be implicated in tumorigenesis through splicing regulation (Yan, 2015).
This study describes the characterization of Nito as a novel component of the Drosophila sex determination pathway. Nito loss-of-function results in stem-cell tumor phenotypes in the germ-line and sexual transformations in the soma. Interestingly, Nito affects Sxl protein levels in both GSCs and somatic tissues by regulating Sxl pre-mRNA alternative splicing, most likely directly as Nito interacts with the Sxl protein and pre-mRNA. The role of Nito is reminiscent of the previously reported roles of splicing factors in Sxl auto regulation, such as both subunits of U2AF, Fl(2)d, SPF45, Vir, and Snf. These data support earlier reports that Sxl physically interacts with components of the spliceosome to simultaneously block utilization of the 3' and 5' splice sites of the male exon (Yan, 2015).
Nito and Spen are members of the SPEN protein family that are evolutionarily conserved from plants, worms, flies to mice and humans. Both proteins contain three N-terminal RRM domains and one C-terminal SPOC domain. The sequence similarity between these domains is low and there is no conservation outside these motifs, suggesting that they have evolved specific functions following a duplication event, as indicated by the observation that spen is not required for Sxl regulation. In Drosophila, spen was first identified in several genetic screens looking for components of the receptor tyrosine kinase (RTK) signaling pathway. Subsequent studies found that spen is implicated in a variety of cellular and developmental processes including neuronal cell fate specification, axon guidance, cell cycle, Hox gene regulation, and cell death. These pleiotropic effects are likely due to the involvement of spen in multiple signaling pathways. However, the molecular mechanisms underlying the function of Spen in these pathways are not understood (Yan, 2015).
Genetic studies in Drosophila have shown that nito overexpression results in a rough eye phenotype and that it plays a redundant role with spen in Wnt signaling, but how Nito is involved in these processes is not known. Biochemical studies indicate that Nito, like its human ortholog, copurify with the precatalytic spliceosome (complex B). In addition, nito, as well as many other splicing factors, was identified in an RNAi screen for RAS/MAPK signaling components. Consistent with these findings, this study found that nito is required for the alternative splicing of the master sex-determination gene Sxl. Previously, both Spen and Nito were thought to act mainly as transcription factors through their SPOC domains, the current findings however clearly indicate that Nito is involved in mRNA splicing. It is intriguing to note that PPS, another important factor required for Sxl splicing, also has a SPOC domain. Similar to Nito, PPS also forms a complex with Sxl protein and its pre-mRNA. In the future it will be crucial to dissect how different protein domains contribute to the function of SPEN family proteins (Yan, 2015).
Then what is the 'main' role of nito? On one hand, the phenotypes in the sex comb, genitalia and germ line appear specific to Sxl and such phenotypes do not depend on the genetic interaction with other genes in the sex determination pathway. On the other hand, nito clearly has other non-sex-specific functions, as revealed by the lethality, rough eye, and wing phenotype observed in both sexes. Because a null allele of nito is associated with zygotic lethality, the RNAi knockdown approach is a powerful method to reveal sex-related phenotypes. Interestingly, the RNAi screen targeting splicing factors did not identify any new additional sex determination genes, indicating that there are a limited number of genes yet to be identified in this pathway. Finally, intriguingly, three recent studies have identified SPEN and Rbm15 (the mouse and human ortholog of Nito) as factors interacting with Xist, the long noncoding RNA that is essential for dosage compensation in mammals. Clearly, future experiments such as RNA-seq will be necessary to elucidate the mechanism and logic of Nito-mediated signaling events (Yan, 2015).
Rbm15, also known as OTT, was originally identified from infants with acute megakaryoblastic leukemia (AMKL). The t(1, 22) chromosomal translocation results in fusion of RBM15 and MKL1, and the fusion protein is responsible for AMKL development as shown in a mouse model. In addition to this chromosome translocation, recent cancer genome sequencing projects have found that RBM15 and SPEN (also known as SHARP) are mutated in many different types of cancers, such as adenoid cystic carcinomas and bladder cancers. Given that SPEN family proteins are frequently mutated or deleted in cancers, they have been proposed to act as potential tumor suppressors. Studies of Spen and Nito in Drosophila will provide mechanistic insights to understanding of this important family of proteins (Yan, 2015).
N(6)-methyladenosine (m(6)A), the most abundant chemical modification in eukaryotic mRNA, has been implicated in Drosophila sex determination by modifying Sex-lethal (Sxl) pre-mRNA and facilitating its alternative splicing. This study identified a sex determination gene, CG7358, and rename it xio according to its loss-of-function female-to-male transformation phenotype. xio encodes a conserved ubiquitous nuclear protein of unknown function. Xio colocalizes and interacts with all previously known m(6)A writer complex subunits (METTL3, METTL14, Fl(2)d/WTAP, Vir/KIAA1429, and Nito/Rbm15), and loss of xio is associated with phenotypes that resemble other m(6)A factors, such as sexual transformations, Sxl splicing defect, held-out wings, flightless flies, and reduction of m(6)A levels. Thus, Xio encodes a member of the m(6)A methyltransferase complex involved in mRNA modification. Since its ortholog ZC3H13 (or KIAA0853) also associates with several m(6)A writer factors, the function of Xio in the m(6)A pathway is likely evolutionarily conserved (Guo, 2018).
The core functions of the insulin/insulin-like signaling and target of rapamycin (IIS/TOR) pathway are nutrient sensing, energy homeostasis, growth, and regulation of stress responses. This pathway is also known to interact directly and indirectly with the sex determination regulatory hierarchy. The IIS/TOR pathway plays a role in directing sexually dimorphic traits, including dimorphism of growth, metabolism, stress and behavior. To understand the degree to which the environmentally responsive insulin signaling pathway contributes to sexual dimorphism of gene expression, the effect of perturbation of the pathway on gene expression was examined in male and female Drosophila heads. The data reveal a large effect of insulin signaling on gene expression, with greater than 50% of genes examined changing expression. Males and females have a shared gene expression response to knock-down of InR function, with significant enrichment for pathways involved in metabolism. Perturbation of insulin signaling has a greater impact on gene expression in males, with more genes changing expression and with gene expression differences of larger magnitude. Primarily as a consequence of the response in males, this study found that reduced insulin signaling results in a striking increase in sex-differential expression. This includes sex-differences in expression of immune, defense and stress response genes, genes involved in modulating reproductive behavior, genes linking insulin signaling and ageing, and in the insulin signaling pathway itself. These results demonstrate that perturbation of insulin signaling results in thousands of genes displaying sex differences in expression that are not differentially expressed in control conditions. Thus, insulin signaling may play a role in variability of somatic, sex-differential expression. The finding that perturbation of the IIS/TOR pathway results in an altered landscape of sex-differential expression suggests a role of insulin signaling in the physiological underpinnings of trade-offs, sexual conflict and sex differences in expression variability (Graze, 2018).
In Drosophila, male courtship behavior is regulated in large part by the gene fruitless (fru). fru encodes a set of putative transcription factors that promote male sexual behavior by controlling the development of sexually dimorphic neuronal circuitry. Little is known about how Fru proteins function at the level of transcriptional regulation or the role that isoform diversity plays in the formation of a male-specific nervous system. To characterize the roles of sex-specific Fru isoforms in specifying male behavior, this study generated novel isoform-specific mutants and used a genomic approach to identify direct Fru isoform targets during development. All Fru isoforms were shown to directly target genes involved in the development of the nervous system, with individual isoforms exhibiting unique binding specificities. fru behavioral phenotypes are specified by either a single isoform or a combination of isoforms. Finally, the utility of these data for the identification of novel sexually dimorphic genomic enhancers and novel downstream regulators of male sexual behavior is illustrated in this study. These findings suggest that Fru isoform diversity facilitates both redundancy and specificity in gene expression, and that the regulation of neuronal developmental genes may be the most ancient and conserved role of fru in the specification of a male-specific nervous system (Neville, 2014).
This study of fru isoform function exemplifies how complex behaviors involved in courtship can be controlled by a single locus. Differential expression of multiple isoforms with different binding specificities produces a 'neural code' of downstream gene expression, in which phenotypes can be specified by either a single isoform or a combination of isoforms. The DamID approach allowed identification of the association of FruM proteins to specific regions of the genome and relate this binding with downstream target genes. Genes known to play a role in the development of the nervous system are significantly overrepresented within these identified FruM target genes. This is certainly consistent with the established role that fru plays in the development of a number of neuronal structures. However, until this study, the identity of fru-regulated genes had not been determined. The identification of putative FruM binding motifs, the strategy for identifying and characterizing Fru-regulated genomic enhancers, and the production of a comprehensive set of fruM isoform-specific mutant flies facilitates an unprecedented leap forward in the ability to study FruM transcriptional regulation (Neville, 2014).
For a more in-depth analysis, concentration was placed on a subset of putative FruMB target genes. The identification of a putative FruMB DNA-binding motif allowed this study to show that the majority of genomic enhancers containing this motif exhibit sexually dimorphic expression in fru neurons. Among the genes associated with these enhancers are the related BTB-Zn-finger genes lola and chinmo, both key neuronal morphogenesis genes. In addition, decreased expression of lola, specifically in fru neurons, led to dramatically reduced levels of male sexual behavior, establishing the necessity of this protein in fru neurons. Since Fru targets other BTB-Zn-finger genes, it is speculated that regulatory diversity of these transcription factors contributes to a neuron-specific transcriptional code leading to specific developmental outcomes. Future examination of these and other FruM target genes will allow deciphering of this code and connect specific dimorphic neural cell fates with behavioral outputs (Neville, 2014).
It was determined that there is a great deal of overlap in the genomic loci targeted by all of the FruM isoforms when it comes to genes involved in the development of the nervous system. Since each Fru isoform appears to have unique binding specificity, it follows that FruM isoforms could act independently on the same genes, either cooperatively or redundantly. FruMB and FruMC isoforms can associate with the same genomic regions containing the putative FruMB motif (see Motif Analysis of FruM Data Sets). FruMB exhibits the most consistent binding specificity, which this study determined to be dependent on amino acid residues that are required for DNA binding. In contrast, although the FruMC isoform is enriched for the FruMB motif, it appears to have a unique DNA binding specificity. Previous analysis of serotonergic neurons in the abdominal ganglion that innervate the male reproductive organs showed evidence of cooperative function between the FruMB and FruMC isoforms, as both were required for the development of these neurons. However, other functions appeared to be isoform specific: for example, only FruMC controls the innervation and formation of the male-specific muscle of Lawrence. Although the elimination of individual FruM isoforms generated overt behavioral deficits, they were not sufficient to abrogate courtship behaviors completely, suggesting some degree of redundancy in the determination of the neural networks directing these behaviors (Neville, 2014).
Alternatively spliced isoforms, like gene duplications, enable a diversification of gene function, by allowing essential (often ancestral) functions to be maintained while others are able to diverge and take on new roles. Evolutionary analysis of the fru C2H2 Zn-finger domains in various insect species shows the appearance and disappearance of these domains throughout evolution. However, the high conservation between all fru C2H2 Zn fingers supports the idea that they all originated from one or a few ancestral sequences and retained a common function (Neville, 2014).
The results support this scenario of evolution in fru, as evidence has been found for both conservation and divergence in function of the different isoforms. Loss of either FruMB or FruMC expression significantly disrupts the male's ability to perform courtship behavior, whereas loss of the FruMA isoform has no obvious consequence. Expression analysis of the individual isoforms in the CNS also mirrors these relationships. FruMB and FruMC isoforms are broadly expressed in most FruM-positive neurons, whereas FruMA expression is restricted to only a subset of FruM-positive neurons. The lack of an overt phenotype associated with the fruΔA mutant may be a reflection of the relative involvement of this subset of fru-positive neurons, or may indicate that the FruMA isoform fulfills more specialized nonessential functions. Evidence has been recently found of positive selection acting on fru exon A across Drosophila species, whereas exons B and C were found to be conserved, supporting the involvement of transcripts containing the A exon in nonessential functions, which may contribute to phenotypic differences between species (Neville, 2014).
Previous microarray experiments showed that genes regulated downstream of FruM (either directly or indirectly) appear to also be regulated by ecdysone. In addition, the ecdysone receptor was shown to act in fru neurons to mediate male courtship behavior. More recently, it was shown that females depleted in ecdysone display male-like courtship behaviors, and it was proposed that distinct ecdysone peaks might regulate the formation of distinct FruM-containing chromatin regulatory complexes. Although the developmental time course did not detect a dramatic shift in FruM DNA-binding specificity as a result of ecdysone pulses, there are more subtle dynamic shifts in binding throughout development that might result from these pulses. A small but significant enrichment was found of known ecdysone-responsive genes in all FruM data sets. Interestingly, this included the cell death gene reaper (rpr), which was identified as a putative target of all FruM isoforms throughout development. fru has been shown to be essential for the suppression of cell death in the male mAL neural cluster, potentially by downregulating key cell death genes. Direct targeting of rpr by FruM isoforms would support this mechanism. This study also identified the ecdysone-responsive transcription factor crooked legs (crol) as a putative target of both FruMB and FruMC isoforms in pupal and adult stages. A previous microarray analysis reported crol as being upregulated in the CNS of fruM mutant males. Deficiency combinations resulting in complete loss of the fru locus (along with a small number of neighboring genes) result in early pupal developmental arrest, around the time of pupal ecdysis. It was noted that the phenotype of fru-deficient flies was similar to that of flies mutant for the ecdysone receptor and the crol gene. Therefore, some of these targets may link the sex-specific and common isoform functions of Fru in response to ecdysone (Neville, 2014).
The identified FruM targets overlap with those of the other key sex-determination protein, Doublesex (Dsx). The male-specific form of dsx (dsxM) is expressed in far fewer cells in the adult CNS than fru, but almost all dsxM cells coexpress fru. Dsx and Fru are the only identified factors at the bottom of the sex-determination hierarchy, and both of these transcriptional regulators act in the same neurons to bring about male-specific neuronal wiring and male-specific behavioral patterns. Given the overrepresentation of identified Dsx target genes in the FruM data sets, it is speculated that FruM and dsxM act together, either in a physical complex or through coregulation of genomic targets, to determine the male-specific nervous system (Neville, 2014).
In the brain of holometabolous insects such as the fruit fly Drosophila melanogaster, the fruitless gene produces sex-specific gene products under the control of the sex-specific splicing cascade and contributes to the formation of the sexually dimorphic circuits. Similar sex-specific gene products of fruitless homolog have been identified in other holometabolous insects such as the mosquitos and the parasitic wasp, suggesting the fruitless-dependent neural sex-determination system is widely conserved among holometabolous insects. However, it remains obscure whether the fruitless-dependent neural sex-determination system is present in basal hemimetabolous insects. To address this issue, the identification, characterization, and expression analyses of the fruitless homolog were conducted in the two-spotted cricket Gryllus bimaculatus as a model hemimetabolous insect. Gryllus fruitless gene encodes multiple isoforms with unique zinc finger domain, and does not encode a sex-specific gene product. Gryllus Fruitless protein is broadly expressed in the neurons and glial cells in the brain, and there was no prominent sex-related difference in the expression levels of Gryllus fruitless isoforms. The results suggest that the Gryllus fruitless gene is not involved in the neural sex-determination in the cricket brain (Watanabe, 2019).
For sexually reproducing organisms, production of male or female gametes depends on specifying the correct sexual identity in the germline. In D. melanogaster, Sex lethal (Sxl) is the key gene that controls sex determination in both the soma and the germline, but how it does so in the germline is unknown, other than that it is different than in the soma. An RNA expression profiling experiment was conducted to identify direct and indirect germline targets of Sxl specifically in the undifferentiated germline. In these cells, Sxl loss does not lead to a global masculinization observed at the whole-genome level. In contrast, Sxl appears to affect a discrete set of genes required in the male germline, such as Phf7. Tudor domain containing protein 5-like (Tdrd5l) was identified as a target for Sxl regulation that is important for male germline identity. Tdrd5l is repressed by Sxl in female germ cells, but is highly expressed in male germ cells where it promotes proper male fertility and germline differentiation. Additionally, Tdrd5l localizes to cytoplasmic granules with some characteristics of RNA Processing (P-) Bodies, suggesting that it promotes male identity in the germline by regulating post-transcriptional gene expression (Primus, 2019).
Sex determination is an essential process in sexually reproducing species, as the production of eggs and sperm depends on the sexual identity of the germ cells and somatic cells of the gonad. In some animals, such as the medaka fish and the house fly, the sexual identity of the soma determines the sexual identity of the germline. But in other animals, such as fruit flies and mammals, the intrinsic sex chromosome constitution (XX vs. XY) of the germ cells is also important for proper gametogenesis. In such cases, the 'sex' of the germ cells must match the 'sex' of the soma in order for proper gametogenesis to occur. While studies have revealed a great deal about how sex is determined in the soma, how germline sexual identity is determined by a combination of somatic signals and germline autonomous properties is much less well understood (Primus, 2019).
In Drosophila, somatic sexual identity is determined by the X chromosome number, with two X's activating expression of the key sex determination gene Sex lethal (Sxl), promoting female identity. The Sxl RNA-binding protein initiates an alternative RNA splicing cascade to allow female-specific splicing of transformer (tra) and, subsequently, doublesex (dsx) and fruitless (fru). dsx and fru encode transcription factors that control somatic sexual identity. Sxl is also the key gene controlling autonomous sex determination in the germline, as Sxl is expressed in the germline in females, and loss of Sxl causes female (XX) germ cells to develop as germline ovarian tumors, similar to male (XY) germ cells transplanted into a female soma. Further, expression of Sxl is sufficient to allow XY germ cells to make eggs when transplanted into a female soma. However, how Sxl is activated in the female germline and how it regulates female germline identity remain unknown, except for the fact that both are different than in the soma. To understand how Sxl promotes female germ cell identity, it is essential to discover its targets in the germline (Primus, 2019).
This work reports an RNA expression profiling (RNA-seq) experiment conducted to identify genes regulated downstream of Sxl in the germline. A previously uncharacterized tudor domain containing protein, Tudor domain protein 5-like (Tdrd5l), was found to be a target of Sxl in the germline. Tdrd5l is strongly expressed in the Drosophila early male germline and is repressed by Sxl activity in the early female germline. It promotes male identity in the germline, and its loss results in germline maintenance and differentiation defects in males, thus reducing their fertility. Tdrd5l protein localizes to cytoplasmic granules related to RNA Processing (P-) Bodies, suggestive of a function in post-transcriptional regulation of gene expression (Primus, 2019).
It has been known for many years that Sxl is necessary for female germline identity, and Sxl has also been shown to be sufficient to allow XY germ cells to undergo oogenesis. It is likely that Sxl plays multiple roles in the germline, both to promote female identity in the early germline, perhaps as early as in the embryonic germline, and in regulating the differentiation of the germline during oogenesis. This study examined the role of Sxl more specifically in the undifferentiated germline through the use of bam mutants. Principle component analysis indicated that, under these conditions, samples with Sxl function reduced in the germline clustered close to control female samples, and far from male samples. While some of the male/female differences may be contributed by the somatic cells present in these samples, it is concluded that reducing Sxl function in the undifferentiated germline does not lead to a dramatic masculinization at the whole-genome level. In contrast, it is proposed that the role for Sxl in the early germline may be restricted to a relatively small number of changes in sex-specific germline gene expression that are important for female vs. male germline function (Primus, 2019).
Recently, a genomic analysis of ovaries mutant for the RNA splicing factor sans fille (snf) was conducted. This is considered to also be a Sxl germline loss-of-function condition as one important change in snf-mutant ovaries is a loss of Sxl expression and an ovarian tumor phenotype that can be rescued by Sxl expression. In contrast to the current results, an increased expression of spermatogenesis genes was observed in snf tumorous ovaries compared to wild type ovaries. It is likely that changes in these 'differentiation' genes were not observed in bam-mutant samples since germline differentiation is arrested at an earlier stage in bam mutants, allowing this study to focus on the undifferentiated germline. Thus, these two analyses can help separate the role of Sxl in regulating early germline sexual identity vs. later aspects of sex-specific germline differentiation. Interestingly, one 'differentiation' gene was identified in both RNA-seq analyses: the testis-specific basal transcription factor TATA Protein Associated Factor 12L (Taf12L or rye). This may indicate that Taf12L could play a role in the undifferentiated germline as well as the later stages of spermatocyte differentiation. In addition, both analyses found evidence for differential regulation of the important male germline identity factor Phf7 (Yang, 2012), where an upstream promoter is utilized preferentially in the male germline and is repressed downstream of Sxl in females. This indicates a role for Phf7 in both the early and differentiating germline. Finally, strong candidates were not observed for targets of alternative RNA splicing regulated by Sxl. The only strong candidate for alternative RNA splicing was the Sxl RNA itself, where the male-specific exon was retained in the residual Sxl RNA from the Sxl RNAi samples. This provides further evidence that Sxl autoregulation occurs in the germline as it does in the soma, as has previously been proposed. It is likely that Sxl may also act at the level of translational control in the germline, as evidence indicates in this study for regulation of Tdrd5l. Future experiments to identify Sxl-associated germline RNAs will be important for investigating this mechanism of action, as has recently been conducted (Primus, 2019).
In addition to its role in sex determination in the soma, Sxl also acts to initiate global X chromosome gene regulation and dosage compensation through translational control of male-specific lethal-2, and it is possible that Sxl plays a similar role in the germline. Whether or not the germline even undergoes dosage compensation is controversial, and thoughtful work has led to opposite conclusions. Further, if dosage compensation does exist in the germline, it must utilize a separate mechanism from the soma, as the somatic dosage compensation complex members msl1 and msl2 are not required in the germline. However, Sxl could retain an msl-independent role to regulate global X chromosome gene expression in the germline. No evidence was observed for this. First, few X chromosome genes were differentially expressed between Sxl- and control samples (30 genes or 1.2% of X chromosome genes tested). Second, the ratio of average gene expression between X chromsome genes and autosomes was very similar in Sxl- females compared to control females (X/A for controls: 1.24, Sxl-: 1.19) and this was similar when considering only genes in particular expression categories (e.g., all genes with some expression in both samples). Thus, it appears likely that Sxl's role in the germline is distinct from that in the soma; it acts to control sex-specific gene regulation and sexual identity in both the germline and the soma, but acts as a general regulator of X chromosome gene expression and dosage compensation only in the soma (Primus, 2019).
Tdrd5l is both a target for Sxl regulation and is important for male germline identity and spermatogenesis. Tdrd5l expression is highly male-biased, both at the RNA and protein levels. When female germ cells are sensitized by partial loss of female sex determination genes, expression of Tdrd5l exacerbates the masculinized phenotype in these germ cells. Significantly, expression of Tdrd5l is sufficient to promote spermatogenesis in XX germ cells present in a male soma (XX tra-mutant testes). Thus, Tdrd5l clearly has a role in promoting male identity autonomously in the germline, which must coordinate with non-autonomous influences from the soma for proper germline sex determination (Primus, 2019).
Loss of Tdrd5l also has a strong effect on male fecundity, even though it is not absolutely required for spermatogenesis. The 50% reduction in fecundity is a strong effect and indicates that Tdrd5l plays an important role in the male germline. However, the fact that some spermatogenesis still proceeds suggests that other factors act in combination with Tdrd5l to control this process. One good candidate is Phf7 which was previously demonstrated to have a similarly important, but not absolute, requirement for spermatogenesis. However, analyses of Tdrd5l, Phf7 double mutants did not reveal any synergistic effect on male germline development. This suggests that other important players in promoting male germline identity remain to be identified (Primus, 2019).
The data indicate that Tdrd5l regulates male germline identity by influencing post-transcriptional gene regulation. Other tudor-domain containing proteins have been shown to act in RNA-protein bodies to influence RNA stability and translational regulation. Further, Tdrd5l localizes to cytoplasmic punctae, specifically a subset of the punctae that also contain Decapping Protein1 (DCP-1), suggesting that these bodies are related to Processing bodes (P-bodies) that are known to control post-transcriptional gene regulation. Interestingly, Tdrd5l's closest homologs, Tejas in flies and TDRD5 in mice, have been shown to regulate piRNA production and transposon regulation. Further, their localization to VASA-containing nuage is thought to influence transposon control. However, no changes were observed in transposon expression regulated by Tdrd5l, and Tdrd5l does not co-localize with VASA in nuage. No genetic interaction was observed between tejas and Tdrd5l. Thus, it is proposed that Tdrd5l plays a distinct role in regulating male germline identity and spermatogenesis, and this may be in the regulation of mRNAs rather than transposons. One possibility is that the role of mouse TDRD5 in transposon regulation and spermatogenesis has been separated into the roles of Tejas in transposon regulation and Tdrd5l in regulating male identity and spermatogenesis in flies. It is widely known that regulation of germline identity is dependent on post-transcriptional mechanisms involving tudor-domain proteins such as the original Tudor protein, which helps define the germ plasm, an RNA body that regulates germline identity. Further, the regulation of sex-specific gametogenesis is also dependent on RNA bodies and their requisite tudor-domain proteins, such as TDRD5 in mouse. These studies indicate that initial germline sexual identity is similarly regulated by post-transcriptional mechanisms, including RNA bodies containing other, distinct, TUDOR-domain proteins such as Tdrd5l (Primus, 2019).
a name="Mercer"> In Drosophila ovaries, germ cells differentiate through several stages of cyst development before entering meiosis. This early differentiation program depends on both the stepwise deployment of specific regulatory mechanisms and on maintenance of germline sexual identity. The study of female sterile mutations that result in formation of germ cell tumors has been invaluable in identifying the mechanisms that control these developmental events. This study characterized the germ cell-enriched gene bourbon (bbn), null mutants of which cause the formation of a mixture of agametic ovarioles and cystic germ cell tumors. Proteomic analysis found Bbn forms a complex with Ovarian tumor (Otu), a protein previously linked with regulation of the sex determination factor Sex lethal (Sxl), and the Drosophila ortholog of c-Myc binding protein (Mycbp). Loss of Mycbp also results in the formation of cystic germ cell tumors. Bbn promotes the stability of Otu and fosters interactions between Otu and Mycbp. Germ cells from bbn and Mycbp mutants display a loss of Sxl expression specifically in the germline. Transgenic rescue experiments show the bbn sterile phenotype is independent from Sxl splicing defects. Further evidence suggests Otu physically interacts with and promotes Sxl protein stability. This function does not depend on Otu's deubiquitinase activity. Last, this study found the human orthologs of Otu and Mycbp, OTUD4, and MYCBP, also physically interact, suggesting conservation of function. Together these data provide insights into how a conserved complex promotes the germline expression of Sxl protein and the differentiation of Drosophila germ cells (Mercer, 2025).
The Drosophila ovary has long served as a useful system for studying adult stem cells and the regulation of germline differentiation. Female Drosophila have two ovaries composed of tube-like structures called ovarioles. Germ cell differentiation starts at the tip of each ovariole in a structure called the germarium. Within germaria, germline stem cells (GSCs) reside in a niche formed by cap cells that produce BMP ligands. These ligands act upon receptors on the surface of GSCs and initiate a signal transduction cascade that results in the transcriptional repression of bag-of-marbles (bam), a gene both necessary and sufficient for germ cell differentiation. Germ cells initiate bam expression once they move away from the cap cell niche. Bam protein physically interacts with Benign gonial cell neoplasm (Bgcn), Meiotic P26 (Mei-P26), and Sex lethal (Sxl) and together repress nanos mRNA translation once germ cells have exited the cap cell niche This repressive activity depends on the ability of Sxl to bind to specific elements within the 3'UTR of nanos mRNA. This complex also likely regulates other mRNAs (Mercer, 2025).
Once differentiation has been initiated, germ cell development proceeds through several discrete steps. Characterizing the mechanisms that regulate the stage-specific expression of different factors, including Sxl, will help inform how germ cells prepare to enter meiosis. Sxl and the Nanos interacting protein Pumilio continue to be expressed until the two-cell cyst stage at which time their expression decreases as the expression of Rbfox1 increases. Rbfox1 is a member of a family of RNA-binding proteins that play roles in the regulation of both alterative splicing and mRNA translation. Loss of Rbfox1 prevents female germ cells from entering meiosis and is accompanied by expanded expression of both Sxl and Pum. Further experiments show that Rbfox1 directly binds to pum mRNA and represses its translation. Sxl mRNA has several potential Rbfox1 binding sites within its 3' UTR, but direct binding has not been demonstrated (Mercer, 2025).
In the context of somatic cells, Sxl acts as a master regulator of sex determination. A widespread model has been that Sxl transcription is turned on from an early promotor in response to the ratio of the X chromosome to autosomes. However, more recent results indicate that the maternally provided protein Groucho (Gro) functions to repress Sxl transcription. Only embryos with two X chromosomes accumulate enough activator to overcome Gro-mediated repression and allow for transcription from Sxl PE. Sxl regulates both alternative splicing and mRNA translation of several downstream targets and promotes its own expression through a feed-forward loop. Later in somatic development, when Sxl is transcribed from its maintenance promotor, cells in females expressing Sxl protein will continue to make functional splice variants of Sxl. By contrast, cells in males produce Sxl mRNA that contains its third exon, which carries a premature stop codon, preventing the production of functional Sxl protein (Mercer, 2025).
What promotes Sxl expression in the female germline is less well understood. Factors including ovo, ovarian tumor (otu), sans fille (snf), and fused (fu) have been shown to function upstream of Sxl. For example, snf encodes a splicing factor and specific mutations in this gene result in loss of female Sxl isoforms specifically in the germline. Ovo promotes the transcription of otu), the founding member of a family of deubiquitinases. In addition to Ovo transcriptional regulation of otu, unknown signals downstream of the female isoform of double sex (dsx) in the soma also promote the expression of otu in the germline. Previous results have shown that otu mutants exhibit misregulation of Sxl splicing, resulting in the production of male-specific isoforms in ovaries. Given the largely cytoplasmic localization of Otu, this regulation of Sxl splicing may be indirect. In addition, several studies have shown that Sxl protein regulates the correct splicing of its own transcripts, further complicating potential interpretations of these results. Thus, the question of how Otu regulates Sxl expression and whether this regulation is direct or indirect remains unclear (Mercer, 2025).
This study identified two additional factors needed for Otu stability and function within the female germline. Loss of the gene CG14545, which is refered to as bourbon (bbn), results in a cystic tumor phenotype similar to that caused by disruption of Sxl and otu in the germline. Bbn is a small, evolutionarily divergent protein found in Drosophilids that structurally resembles mammalian c-Myc Binding Protein (MYCBP). Biochemical experiments indicate that Bbn protein interacts with both Otu and the actual Drosophila ortholog of MYCBP. Loss of Mycbp results in the formation of cystic germ cell tumors, mimicking the differentiation defects observed in Sxl, otu, and bbn mutants. Bbn promotes the stability of Otu and fosters physical interactions between Otu and Mycbp. Germ cells from bbn and Mycbp mutants, like otu mutants, display a loss of Sxl protein expression specifically in the germline. Otu physically interacts with a germline-specific isoform of Sxl protein and likely protects it from protein degradation. This function does not appear to require the deubiquitinase activity of Otu. These results provide insights into previously uncharacterized posttranslational mechanism that promotes the protein expression of female Sxl isoforms in the Drosophila germline (Mercer, 2025).
This study reports the identification and characterization of a complex composed of Otu and two structurally related proteins: Bbn and Mycbp. otu and bbn mRNA expression appears specific to germ cells while Mycbp displays broader expression across different tissues. Loss of any of these factors causes female sterility, marked by the appearance of agametic ovarioles and cystic germ cell tumors that fail to differentiate beyond the earliest stages of germ cell development. A regulatory target of the Otu/Bbn/Mycbp complex in adult ovaries is the sex determination factor Sxl and expression of Sxl protein, but not mRNA, is lost upon disruption of any members of the complex (Mercer, 2025).
AlphaFold modeling predicts that Bbn and Mycbp interact with Otu in a region away from its catalytic domain and between Otu's deubiquitinase domain and Tudor domain. Biochemical experiments presented in this study appear to support this model. Moreover, Bbn promotes the stability of Otu and is required for interactions between Otu and Mycbp. The common phenotypes exhibited by null mutations in bbn, Mycbp, and otu further support the idea these three proteins form a complex that acts to promote Sxl protein expression in adult germaria. However, this complex likely has other targets in addition to Sxl. bbn, Mycbp, and otu mutants exhibit a partially penetrant agametic phenotype. At least for bbn mutants, this agametic phenotype does not worsen with age, indicating germ cells are not constantly lost in the absence of this complex. Previous gene expression analysis indicates that bbn and otu are first expressed in germ cells during embryogenesis. Together these data suggest that the Otu/Bbn/Mycbp complex plays a role during early germ cell specification, maintenance, or migration. Further analysis will be required to determine when bbn, Mycbp, and otu mutant phenotypes first begin to manifest during germ cell development (Mercer, 2025).
The Bbn and Otu complex may also function at additional steps during late germ cell differentiation. Weak hypomorphs of otu and the HA::bbn allele both display a dumpless phenotype during late oogenesis. Both allele both display HA::bbn and otu weak hypomorphs have nurse cell nuclei that do not decondense properly after the 5 blob stage reminiscent of a cup phenotype. Previous work in the literature has demonstrated that cup and otu genetically interact. Cup was a top hit in the Otu proteomic analysis and was identified in the Bbn proteomic analysis as well. While this study focused on Sxl in this current study, Cup represents an exciting potential regulatory target for future study (Mercer, 2025).
The data support a model in which the Otu/Bbn/Mycbp complex acts downstream of transcription and splicing of Sxl. Sxl splicing defects in bbn mutant ovaries can be rescued by expression of a Sxl cDNA transgene, while the tumorous phenotype of bbn mutants is not. In addition, the localization of Bbn and Otu in the cytoplasm supports the idea that the complex likely promotes Sxl translation or protein stability. Evidence is provided that Sxl is regulated by the proteasome and is potentially ubiquitinated, consistent with previous results. However, Otu's deubiquitinase activity is not required for Sxl stability. Interestingly, proteomic analysis reveals that Otu physically interacts Sxl-PX and Sxl-PY proteins, suggesting that Otu may protect Sxl proteins from degradation in a manner that does not involve deubiquitination. Future experiments should aim to determine how the Otu/Bbn/Mycbp complex carries out this function (Mercer, 2025).
Whether Otu functions as a deubiquitinase has been debated in the literature. Drosophila Otu has a serine in the active site instead of the canonical cysteine. Mutating the D37, S40, and H143 residues all independently result in a loss of deubiquitinase activity in vitro. The current data indicate that the S40 residue is dispensable for early germ cell development and Sxl protein expression. Because the OTU deubiquitinase domain is not required, other domains of Otu may be responsible for promoting Sxl protein stability. Previous structure-function work with Otu demonstrates that Otu's Tudor domain is required for germ cell proliferation and cyst formation in the germarium. Future study of the function of the Otu Tudor domain could bring needed insight into the regulation of Sxl protein and other Bbn, Mycbp, and Otu complex targets (Mercer, 2025).
Last, this study detected interactions between OTUD4, the close human homolog of Drosophila OTU, and MYCBP in human cell lines. There are several plausible distant homologs of Bbn that could be tested, or alternatively, MYCBP could form a dimer in the complex. Regardless, physical interactions between OTUD4 and MYCBP reveal that the association of these proteins has been conserved across evolution. OTUD4 has been shown to function as a deubiquitinase with different substrate specificities. OTUD4 has also been shown to act as a scaffold for other deubiquitinases, thus promoting the protein stability of some targets in a catalytic independent manner. This raises the possibility that there are other interacting partners needed for Sxl protein stability. In addition, OTUD4 also interacts with RNA. Whether Drosophila Otu has similar functions remains to be tested (Mercer, 2025).
Analyses of orthologs of the Drosophila genes identified in non-drosophilid taxa revealed that evolution of sex determination pathways is consistent with a bottom-up mode, where only the terminal genes within the pathway are well conserved. doublesex (dsx), occupying a bottom-most position and encoding sex-specific proteins orchestrating downstream sexual differentiation processes, is an ancient sex-determining gene present in all studied species. With the exception of lepidopterans, its female-specific splicing is known to be regulated by transformer (tra) and its co-factor transformer-2 (tra2). This study shows that in the African malaria mosquito Anopheles gambiae, a gene, which likely arose in the Anopheles lineage and which was called femaleless (fle), controls sex determination in females by regulating splicing of dsx and fruitless (fru; another terminal gene within a branch of the sex determination pathway). Moreover, fle represents a novel molecular link between the sex determination and dosage compensation pathways. It is necessary to suppress activation of dosage compensation in females, as demonstrated by the significant upregulation of the female X chromosome genes and a correlated female-specific lethality, but no negative effect on males, in response to fle knockdown. This unexpected property, combined with a high level of conservation in sequence and function in anopheline mosquitoes, makes fle an excellent target for genetic control of all major vectors of human malaria (Krzywinska, 2021).
Dosage compensation, the balancing of X-linked gene expression between sexes and to the autosomes, is critical to an organism's fitness and survival. In Drosophila, dosage compensation involves hypertranscription of the male X chromosome. This study used quantitative live imaging and modeling at single-cell resolution to study X chromosome dosage compensation in Drosophila. The four X chromosome genes studied undergo transcriptional bursting in male and female embryos. Mechanistically, the data reveal that transcriptional upregulation of male X chromosome genes is primarily mediated by a higher RNA polymerase II initiation rate and burst amplitude across the expression domain. In contrast, burst frequency is spatially modulated in nuclei within the expression domain in response to different transcription factor concentrations to tune the transcriptional response. Together, these data show how the local and global regulation of distinct burst parameters can establish the complex transcriptional outputs underpinning developmental patterning (Beadle, 2023).
MSL2, the DNA-binding subunit of the Drosophila dosage compensation complex, cooperates with the ubiquitous protein CLAMP to bind MSL recognition elements (MREs) on the X chromosome. This study explored the nature of the cooperative binding to these GA-rich, composite sequence elements in reconstituted naive embryonic chromatin. It was found that the cooperativity requires physical interaction between both proteins. Remarkably, disruption of this interaction does not lead to indirect, nucleosome-mediated cooperativity as expected, but to competition. The protein interaction apparently not only increases the affinity for composite binding sites, but also locks both proteins in a defined dimeric state that prevents competition. High Affinity Sites of MSL2 on the X chromosome contain variable numbers of MREs. The cooperation between MSL2/CLAMP was not influenced by MRE clustering or arrangement, but happens largely at the level of individual MREs. The sites where MSL2/CLAMP bind strongly in vitro locate to all chromosomes and show little overlap to an expanded set of X-chromosomal MSL2 in vivo binding sites generated by CUT&RUN. Apparently, the intrinsic MSL2/CLAMP cooperativity is limited to a small selection of potential sites in vivo. This restriction must be due to components missing in the reconstitution, such as roX2 lncRNA (Eggers, 2023).
The experimental strategy of this study involves the assembly of physiological chromatin on genomic DNA to provide a complex substrate for TF interaction assays. Since the assembly system is derived from Drosophila preblastoderm embryos, the chromatin that arises has all hallmarks of pre-MBT embryos: it does not contain significant concentrations of endogenous TFs or components of the transcription machinery. The addition of defined TFs somewhat mimics aspects of zygotic genome activation, when newly translated proteins initiate the complex genome expression program. The analysis of large compendia of potential TF binding sites in different genomic contexts allows statistical generalization, in addition to anecdotal illustration (Eggers, 2023).
This experimental system is well suited to discovering intrinsic protein-DNA interactions of well-characterized components. These may even include heterologous TFs of other species that bind short recognition motifs statistically distributed in Drosophila genomes. Characterizing the intrinsic binding properties of TFs in a complex chromatin environment is important, even if TF binding specificities commonly observed in vitro do not explain physiological chromosome interactions (Eggers, 2023).
The focus of this study has been on the DNA binding proteins that target the 'male-specific-lethal' (MSL) dosage compensation complex (DCC) exclusively to X chromosome-specific DNA elements to deploy the vital activation of transcription. In vivo, the MSL proteins do not bind autosomal DNA. This study found that (i) MSL2, the DNA-binding subunit of the DCC, bears intrinsic sequence selectivity for a complex X chromosome-specific DNA signature, which is termed PionX sites; (ii) MSL2 cooperates with the abundant GA-binding protein CLAMP to associate with bona fide high affinity sites (HAS) on the X chromosome in vivo. In vitro this cooperation results in the recruitment of MSL2 to hundreds of strong CLAMP binding sites on all chromosomes; (iii) the GA-binding GAF competes with CLAMP/MSL2, preventing binding to a class of unphysiological sites. This study investigated the mechanism underlying the CLAMP-MSL2 cooperativity (Eggers, 2023).
The cooperativity between CLAMP and MSL2 was found to depend on the physical interaction between the two proteins. Although a protein complex is formed in solution, it is possible or even likely that the interaction is promoted by DNA. It is expected that upon disruption of the TF interaction the direct cooperativity would turn into indirect, nucleosome-mediated cooperativity, the dominant mechanism of synergism between TFs. Surprisingly, it was found that under those conditions, the stronger GA-binder, CLAMP, competes with MSL2 for binding to composite sites. These sites are unusually long: MEME detects PWMs of 20 bp and more in peaks, so there should be space for two or more proteins to bind side by side. This is in line with the comprehensive study of Taipale and colleagues, who showed that often cooperating TFs bind composite sequence elements where individual TF recognition motifs overlap (Jolma, 2015). These findings argue that the physical interaction not just serves to provide additional surfaces to reduce the off-rate of TFs, but in this case assures that the two cooperating factors bind DNA in a defined arrangement that assures that the long, composite binding sites are fully used and to prevent dominance by the more avid GA-binder CLAMP, which outcompetes MSL2 if not physically connected to it (Eggers, 2023).
Since CLAMP binds degenerate GA repeats, it is possible that two CLAMP molecules can bind to these long sequences, or that single CLAMP molecules bind with variable translational positions, thus occluding GA sequences required for MSL2 interaction. Accordingly, MSL2 can resist competition at sites that contain the PionX signature, but will be competed away from sites of lower affinity that contain degenerate GA repeats. In such a situation, the physical interaction between CLAMP ZF1 and the CBD of MSL2 may ensure that both proteins lock into a defined geometry that prevents competition (Eggers, 2023).
MSL2 and CLAMP mutually cooperate to bind stably to X-chromosomal HAS in vivo. However, the chromosomal MSL2 binding profile only overlaps to a low extent with the pattern of MSL2-CLAMP binding in vitro. In some ways, this is not surprising given that MSL2 functions as a subunit of the large DCC. It is possible that MSL1-induced dimerization improves the selectivity of DNA recognition. The DCC also contains the long, non-coding roX RNA. Indeed, several studies concluded that the presence of roX RNA is required for faithful localization of MSL2 on the X chromosome. This study found MSL2 delocalized to strong CLAMP binding sites on all chromosomes in the absence of roX RNA in vivo. Although the underlying mechanism is unknown, it is noted that roX2 interacts with the C-terminus of MSL2, which also harbors the intrinsic GA-binding specificity of MSL2 as well as the CBD. It was speculated that roX may modulate the DNA and protein interactions of MSL2, perhaps by restricting the cooperativity with CLAMP (Eggers, 2023).
Remarkably, it has been recently observed that at nuclear division cycle 13, prior to the expression of roX2 and the assembly of a mature DCC, MSL2 and CLAMP localize to CLAMP binding sites on all chromosomes. This study hypothesized that the binding profile in reconstituted preblastoderm chromatin might reflect some of these earliest MSL2 interactions with chromosomes prior to roX2 expression. However, a comparison of the profiles did not support this hypothesis (Eggers, 2023).
Zygotic gene activation is also accompanied by the appearance of the linker histone H1, which may modulate TF cooperativity. This possibility was explored in earlier work, but it was found found that genome-wide incorporation of H1 did not affect the cooperativity of CLAMP and MSL2 qualitatively, but led to a general dampening of all TF binding to chromatin (Eggers, 2023).
Taking advantage of the superior resolution and signal/noise output of C&R analyses determined MSL2 binding sites in S2 cells. More that 700 distinct peaks of MSL2 interaction was obtained, all of which were localized on the X chromosome. Stratifying the peaks according to their signal strength reveals a hierarchy of sequences. Motifs with 5' extension, resembling the PionX motif ranged among the sites with highest affinity, followed by sites of intermediate affinity that display the MRE consensus sequence. Weaker binding sites contain degenerate MRE motifs dominated by GA repeats. These data support earlier conclusions from low-resolution studies on polytene chromosomes about the existence of a hierarchy of binding sites, where sites of higher affinity are primary attractants for MSL proteins and sites of lower affinity profit from local TF enrichment (Eggers, 2023).
The X-chromosomal HAS or 'chromosome entry sites, CES' were initially defined at low resolution on polytene chromosomes. With the advent of the ChIP-seq methodology it became clear that the peaks of DCC binding are several hundred base pairs long and often contain two or more MRE sequences. MEME analysis often focuses only on the strongest MRE within a larger ChIP-seq peak. Intuitively, loci with multiple MREs may attract more MSL2 than sites with a single MRE. It is also conceivable that higher affinity sites have a defined arrangement of sites, which may facilitate cooperative interactions between MSL complexes bound to different elements. However, these questions have not been systematically investigated (Eggers, 2023).
With respect to the diversity of MRE number and arrangement, which may or may not include PionX motifs, the intrinsic binding sites obtained in vitro and the physiological X chromosomal binding sites appear very similar. In both cases, no strong correlation was found between the number, length or arrangement of MRE sequences with ChIP-seq peak intensity. It is concluded that the binding strength is mainly determined by the affinity of MSL2 for individual MRE motifs, where the cooperativity with CLAMP plays an important role. The predominant binding event can be inferred from EChO analyses. MSL2 appears particularly dependent on this cooperation, since embryonic nuclei only contain very limited amounts of MSL2, barely enough to distribute one copy for each binding site mapped by C&R (Eggers, 2023).
It has been suggested that MREs originally evolved from pyrimidine-rich splicing enhancers in introns and were not under selection for precise location, but rather for proximity to genes. The finding that MSL2/CLAMP binding does not require a defined architecture of binding sites resonates with the observation that defined spacing and orientation of multiple binding sites are commonly not needed to assure integration of different TF input (Eggers, 2023).
In conclusion, these analyses show that the direct contact between MSL2 and CLAMP ensures cooperativity and avoids competition between the two proteins at individual MREs. Remarkably, in vivo this intrinsic mechanism only operates at X chromosomal sites where the DCC is functional. Future research will focus on finding the molecular mechanism that restricts this cooperative binding to the X chromosome (Eggers, 2023).
Although sex chromosomes have evolved from autosomes, they often have unusual regulatory regimes that are sex- and cell-type-specific such as dosage compensation (DC) and meiotic sex chromosome inactivation (MSCI). The molecular mechanisms and evolutionary forces driving these unique transcriptional programs are critical for genome evolution but have been, in the case of MSCI in Drosophila, subject to continuous debate. This study took advantage of the younger sex chromosomes in D. miranda (XR and the neo-X) to infer how former autosomes acquire sex-chromosome-specific regulatory programs using single-cell and bulk RNA sequencing and ribosome profiling, in a comparative evolutionary context. Contrary to mammals and worms, the X down-regulation through germline progression was found to be most consistent with the shutdown of DC instead of MSCI, resulting in half gene dosage at the end of meiosis for all 3 X's. Moreover, lowly expressed germline and meiotic genes on the neo-X are ancestrally lowly expressed, instead of acquired suppression after sex linkage. For the young neo-X, DC is incomplete across all tissue and cell types and this dosage imbalance is rescued by contributions from Y-linked gametologs which produce transcripts that are translated to compensate both gene and protein dosage. An excess was found of previously autosomal testis genes becoming Y-specific, showing that the neo-Y and its masculinization likely resolve sexual antagonism. Multicopy neo-sex genes are predominantly expressed during meiotic stages of spermatogenesis, consistent with their amplification being driven to interfere with mendelian segregation. Altogether, this study reveals germline regulation of evolving sex chromosomes and elucidates the consequences these unique regulatory mechanisms have on the evolution of sex chromosome architecture (Wei, 2024).
The status of meiotic sex chromosome regulation in Drosophila has been debated for decades. The first evidence for MSCI in flies, albeit indirect, was based on observations of reciprocal translocations between the X and autosome. Autosome-to-X translocations cause male sterility but not X-to-autosome translocations, suggesting that germline-essential genes translocated onto the X are down-regulated. In conjunction with claims of increased X-condensation in early spermatocytes, this led to the suggestion of MSCI in flies but the cytological data were subsequently refuted. More recent, direct evidence of MSCI includes reduced expression of transgenes inserted onto the X versus autosomes, and reduced expression of X-linked genes in carefully dissected testes tissues as assayed by qPCR and microarrays. In conjunction with the observation that genes with testes-biased expression are underrepresented on the X in flies, it has been proposed that MSCI drives the exodus of testes-biased genes off the X chromosome onto autosomes. However, others have argued that the X is not globally inactivated, or that the reduced expression is not exclusive to meiotic stages but occurs even in premeiotic cells. Discrepancies have been attributed to limitations in methodologies including limited number of genes quantified and tissue contamination in manual dissections, issues that scRNA-seq holds the promise of resolving. When analyzing the meiotic cell types in adult Drosophila testes scRNA-seq, Establishing the presence of MSCI, reporting spermatocyte expression levels that are 'expected from incomplete or absent dosage compensation' was not possible in one study. However, another srudy reported the presence of MSCI after analyzing scRNA-seq generated from developing testis discs in male larva, although the reported down-regulation does not exceed half of autosomal expression (and thus is also consistent with absence of DC in testes). Therefore, even with scRNA-seq relieving concerns of cell-type contamination the status of MSCI in Drosophila remains debated (Wei, 2024).
Advantage of D. miranda's unique sex chromosome architecture to evaluate the presence of MSCI in conjunction with scRNA-seq. D. miranda's 3 X chromosome arms of different ages allowed determination of whether and how the X chromosome is down-regulated during spermatogenesis, and how it evolves. Inconsistent with the X being inactivated during meiosis, expression of X-linked genes increases through meiosis, although to a lesser extent compared to the autosomes. Furthermore, overall expression of the X's never drops below half that of the autosomes. Contrasting expression trajectories of the different X chromosomes is particularly diagnostic as to what process influences X expression during spermatogenesis, since DC shutdown and MSCI make different predictions for X arms with different ages. If the X is silenced by MSCI, the neo-X is expected to be the least down-regulated due to its most recent autosomal heritage, while under DC shutoff expression of all 3 X's is expected to simply equalize (but not drop below half of autosomal expression levels). Prior to meiosis, the neo-X consistently has the lowest expression of all 3 X's, indicating incomplete dosage compensation. However, this study found that the neo-X loses expression the quickest and through meiosis, expression of older X's simply drops to comparable levels. Expression levels of all 3 X arms equalize after meiotic exit at roughly 58.5% of autosomal expression. These results collectively argue that the X's are losing dosage compensation during spermatogenesis, resulting in relatively lower expression compared to autosomes. One critical benefit of examining the neo-X in D. miranda is the possibility of inferring the ancestral expression prior to sex linkage by analyzing autosomal orthologs in D. pseudoobscura and neo-Y linked gametologs. Both comparisons revealed that low expression of a subset of neo-X genes most likely reflects ancestrally low expression, instead of acquired repression after X-linkage (Wei, 2024).
This model of relative X down-regulation through DC loss reconciles discrepancies in several studies. It is fully consistent with both the scRNA-seq findings in in previous studies. In one study, cytological data showing reduced active RNA-Polymerase-II (Pol-II) staining in meiotic nuclei was argued as critical support for MSCI-driven down-regulation. In light of the current results, it is suspected that reduced Pol-II activity reflects the attenuation or removal of DC. MSL-associated hyper-transcription in somatic cells is achieved via elevated levels of Pol-II recruitment to promoters and enhanced elongation. The cytological results in a previous study showed that active Pol-II signals on the X, estimated as the ratio between active and total Pol-II signals, appear to be around half of the ratio for autosomes, suggesting that half of the Pol-II occupying the X are no longer active. While this was interpreted as MSCI, it could also represent reduction of Pol-II activity on the hyper-transcribed X without clearing Pol-II occupancy, and therefore is fully consistent with DC shutdown. Moreover, their volumetric estimates suggest that the X is less compacted and occupies more nuclear space than autosomes; this is inconsistent with the compaction of the X as expected of and observed in animals with MSCI. Interestingly, a recent study profiled active Pol-II in spermatocytes of meiotic arrest mutants and reported that genome-wide active Pol-II profiles across the X and autosomes are fully consistent with a lack of dosage compensation by this stage of spermatogenesis. No evidence of silencing chromatin enrichment on the X was found in spermatocytes and it was similarly concluded the lack of DC as the cause of X down-regulation. The current results are also in partial agreement with the reports in another study that down-regulation of the X occurs across the testes and is not specific to meiosis, as the X:A expression ratios in somatic (Cyst and Hub) and early germline clusters (GSC and SG) are all <1 and thus lower than expected under full dosage compensation (Wei, 2024).
These analyses comparing X-linked genes proximal and distal to the MSL complex clearly shows DC up-regulating the former in many of the testis clusters until late meiotic cell types (late spermatocytes). While this is consistent with previous reports in D. melanogaster testes scRNA-seq dataset, the germline is thought to lack MSL-dependent dosage compensation in flies. This is primarily based on the cytological observation that many of the MSL components are absent in the germline, the male germline appears to lack X-chromosome-specific H4K16ac and MSL-1 and MSL-2 are dispensable for spermatogenesis in pole cell transplants. However, this study found that only roX1 and mof are absent or lowly expressed in the germline tissues of D. miranda. Rox1 and rox2 have essential but redundant function in dosage compensation and mof, the histone acetyltransferase, appears to be lowly expressed even in other scRNA-seq datasets for somatic tissues that are expected to be dosage compensated, so low expression of mof may not necessarily reflect absence of activity. Thus, the data are consistent with MSL-dependent DC in the testis of D. miranda, but alternative means of achieving X up-regulation in the GSC and spermatocytes without hyper-acetylation are possible. Three-strand DNA-RNA-hybrid structure (R-loops) were shown to be associated with hyper-transcription on the X in the absence of H4K16Ac. Further, a recent comparison of the nuclear topology of germline stages reported that in spermatogonia and spermatocytes, active chromatin regions on the X have more intra-chromosomal contacts than the autosomes, potentially suggesting that active regions on the X are gathering at transcription-rich nuclear territories allowing for higher expression. These studies both suggest alternative mechanisms of up-regulating the X in the absence of the full MSL complex (Wei, 2024).
Curiously, the (perhaps, non-canonical) dosage compensation in the germline appears to differentially up-regulate the X arms. The neo-X shows incomplete dosage compensation across all tissues, even somatic ones. However, incomplete dosage compensation becomes much more pronounced in the early germline, where the extent of dosage compensation tracks the age of the X's. This suggests that germline DC is either less robust or being lost more quickly on the younger neo-X (Wei, 2024).
While dosage compensation of the neo-X appears most incomplete in testis, this study`also found that expression of the neo-Y alleles can increase overall expression from the neo-sex chromosomes. By examining the expression of gametologs on the neo-sex chromosomes in the scRNA-seq and comparing their expression to the ancestral autosomal expression in bulk RNA-seq data, this study found that the neo-Y chromosome can mitigate suboptimal dosage compensation on the neo-X, regardless of the tissue type. Importantly, since neo-Y alleles are more highly expressed in testis compared to most autosomal tissues, this means that overall expression from the neo-sex chromosomes in testis resembles that of the older X chromosomes more closely-i.e., only a moderate deficiency of Muller C linked expression relative to autosomes in testis, similar to that of the older X chromosomes (Wei, 2024).
Gene content of sex chromosomes is non-random, and gene expression evolution and gene movement can contribute to sex chromosome-specific expression patterns. The neo-Y is becoming masculinized. Ancestrally testes-biased genes (inferred from expression patterns in D. pseudoobscura) have become either neo-Y biased in expression or neo-Y exclusive, indicating either down-regulation or complete loss of the neo-X gametologs, respectively. Demasculinization of the X chromosome occurs, in part, due to the fact that the X chromosome spends twice as much time in females than males, and previous studies in Drosophila have revealed an excess of gene movement of testis-expressed genes off the X chromosome onto autosomes. This study showed that the neo-Y presents an intermediary for this movement as male-biased genes first transition to neo-Y biased or specific expression, thereby alleviating the potential for these genes to be female-detrimental and resolving sexual conflict. However, as degeneration continues, these genes will inevitably be lost, further driving their movement onto autosomes. Interestingly, the formation of the neo-X in D. miranda is not associated with an excess of gene movements from Muller C to other autosomes; in fact, gene movement is small and Muller A appears to be the most frequent destination. Thus, autosomal migration likely occurs only after further degeneration of the neo-Y which provides a temporary but unstable solution to sexual conflict (Wei, 2024).
Sex chromosomes are also vulnerable to be involved in conflicts during meiosis. Meiotic drivers that try to cheat fair mendelian segregation are prone to originate on sex chromosomes. Sex chromosome drive will skew the population sex ratio and select for suppressors on the other sex chromosome that are resistant to the distorter. If the distorter and suppressor are dosage sensitive, they would undergo iterated cycles of expansion, resulting in rapid co-amplification of driver and suppressor on the X and Y chromosome. Recently it was shown that meiosis and RNAi-related genes co-amplified on both the neo-X and neo-Y chromosome of D. miranda. Consistent with co-amplification of X/Y genes being driven to interfere with mendelian segregation, this study showed that these genes are predominantly expressed during meiotic stages of spermatogenesis and postmeiotic stages, the cell types in which meiotic drivers are most likely to operate. Thus, sexual and meiotic conflicts may drive unique gene content and expression evolution on sex chromosomes (Wei, 2024).
Germline cells use distinctive variations on transcriptional gene regulation, and studies of Drosophila spermatogenesis have detailed many specialized alterations of core general transcription factors that direct expression programs as differentiation proceeds. However, the chromatin features of gene regulation in spermatogenesis have been less thoroughly characterized, in part because of the complexity of the tissue and limiting numbers of germline cells. This was addressed by performing efficient CUT&Tag chromatin profiling for both active and repressive chromatin marks in the Drosophila testis and in isolated spermatocytes. These profiles reveal several notable features of the epigenome in the differentiating germline. First, integration of chromatin marks with published gene expression data details a general correspondence between gene promoter activation and mRNA production as expected, but for a fraction of genes their promoters activate earlier than expected. Second, genes that are activated late in spermatogenesis tend to have very reduced active chromatin marks at their promoters. Third, while many active genes in somatic cells accumulate RNAPII near their gene starts, such accumulation is absent in spermatocytes. Fourth, integration of chromatin profiling for multiple chromatin marks and profiling of RNAPII demonstrate that the single X chromosome is neither dosage compensated nor globally inactivated in spermatocytes. Finally, histone H2A mono-ubiquitinylation appears to have a specialized role in modulating expression from the heterochromatic Y chromosome (Anderson, 2023).
The uniform distribution of RNAPII throughout active genes in male germline cells is strikingly different from the typical accumulation of RNAPII near promoters in somatic cells. Promoter-proximal accumulation results from dynamic pausing of RNAPII before conversion into the productive elongating isoform, and is a major control point for transcriptional regulation in somatic cells. Thus, the uniformity of RNAPII across expressed genes in spermatocytes suggests that pausing does not occur, necessitating gene regulation solely by transcription factor and RNAPII recruitment. This may allow for a simpler promoter architecture, and indeed spermatogenic gene promoters are distinctively small. Alternatively, RNAPII progression through gene bodies may be slow, altering the steady-state distribution of elongating polymerase. Further, either of these changes in RNAPII behavior would affect chromatin features of active genes. RNAPII binds enzymes that progressively methylate the lysine-4 residue of histone H3, and so active promoters in somatic cells are typically marked with both H3K4-dimethylation and -trimethylation. A consequence of changing the rate of polymerase progression in spermatocytes would be reduced methylation of active promoters (which is most severe in late spermatocyte and post-meiotic stages), and thereby reduced reliance of these marks for promoting transcription (Anderson, 2023).
While protein and lncRNA components of the somatic dosage compensation machinery are not produced in the male germline, transcriptional profiling described up-regulation of the single X chromosome in early germline stages. The mechanism for this non-canonical dosage compensation remains unknown, but by the spermatocyte stage X-linked genes are no longer up-regulated. Profiling of RNAPII confirms that there is no up-regulation of this chromosome in spermatocytes. There has been substantial investigation into whether the X is inactivated in spermatocytes, inspired by the regulation of sex chromosomes in mammals and the moderate depletion of male-germline-expressed genes on the Drosophila X chromosome. In mammalian male germlines the X and Y chromosomes undergo MSCI, where these chromosomes are precociously silenced just before meiosis. A variety of chromatin features accumulate across the sex chromosomes at this time, including the enrichment of both H3K9me2 and ubiquitinated histone H2A (uH2A) histone modifications. However, this study found that neither of these histone modifications is enriched on the X chromosome in the Drosophila male germline. This combined with the equivalent amounts of RNAPII on X-linked genes and autosomal ones implies that there is no X chromosome inactivation in Drosophila (Anderson, 2023).
Why do flies differ from mammals? Mammalian MSCI is considered to be an elaborated response of germline cells to the detection of the unpaired sex chromosomes. However, meiosis in Drosophila males is unusual in that all the chromosomes do not synapse nor recombine. Logically, the evolutionary loss of synapsis must have required the concomitant loss of an unpaired chromosome response in Drosophila males. Indeed, many of the proteins recruited to the mammalian sex body are normally involved in meiotic recombination but have been repurposed for unpaired chromosome inactivation, supporting the idea that MSCI is mechanistically linked to synapsis. Alternatively, mammalian MSCI may be a variant of the X-inactivation system that operates in females for dosage compensation but since Drosophila solved the dosage compensation problem without inactivation, this precluded the evolution of MSCI. These possibilities are not mutually exclusive (Anderson, 2023).
The Y chromosome in Drosophila is unique in that it is almost entirely composed of repetitive sequence, but also carries some unique genes required for male fertility. Thus, in somatic cell types this a heterochromatic chromosome, but is heavily transcribed in spermatocytes. This activated chromosome accumulates multiple histone modifications, some of which have peen profiled in this study. Surprisingly, one of the histone modifications that coats the activated Y chromosome is mono-ubiquitinylation of histone H2A. This modification is typically associated with Polycomb-mediated gene silencing in somatic cells, where it is catalyzed by the Sce/RING1B enzyme subunit of the Polycomb repressive complex 1 (PRC1). However, the Polycomb subunit of PRC1 does not co-localize with the uH2A-coated Y chromosome in spermatocytes, suggesting that it is not catalyzed by Sce here. Indeed, uH2A also coats the X and Y chromosomes in mammalian spermatocytes, where this modification is catalyzed by a distinct enzyme, the UBR2 E3 ubiquitin ligase. The Drosophila genome encodes a homolog of this protein family, as well as many other ubiquitin ligases, some of which target the H2A histone. It is unknown which enzyme is responsible for the uH2A modification in the Drosophila male germline, although some candidates have male sterile phenotypes when mutated. While it is surprising to find a histone modification conventionally associated with silencing enriched on an activated chromosome (and indeed the reason this study profiled it was as a putative MSCI marker), the roles of uH2A in gene regulation are diverse even in somatic cells. The uH2A modification is associated with both silenced and some active genes in developing eye tissue, and counters chromatin compaction in the early embryo. In addition to the Y chromosome, a number of heterochromatic transposons in the Drosophila male germline are also activated and accumulate the uH2A modification. Thus, it is conceivable that this modification - perhaps in combination with other marks - works generally to modulate transcription of extremely heterochromatic regions in spermatocytes (Anderson, 2023).
This study aims at identifying transcriptional targets of FruitlessBM (FruBM), which represents the major isoform of male-specific FruM transcription factors that induce neural sexual dimorphisms. A promoter of the axon-guidance factor gene robo1 carries the 16-bp palindrome motif Pal1, to which FruM binds. A genome-wide search for Pal1-homologous sequences yielded ~200 candidate genes. Among these, CG17716 potentially encodes a transmembrane protein with extracellular immunoglobulin (Ig)-like domains similar to Robo1. Indeed, FruBM overexpression reduced CG17716 mRNA and protein expression. In the fru-expressing mAL neuron cluster exhibiting sexual dimorphism, it was found that CG17716 knockdown in female neurons completely transformed all neurites to the male-type. Conversely, CG17716 overexpression suppressed male-specific midline crossing of fru-expressing sensory axons. CG17716 was renamed teiresias (tei) based on this feminizing function. It is hypothesized that Tei interacts with other Ig superfamily transmembrane proteins, including Robo1, to feminize the neurite patterns in females, whereas FruBM represses tei transcription in males (Sato, 2020).
Reproductive success in many male animals relies on their behavioral performance during mating attempts with a female1. The neural circuitry that controls mating behavior has thus evolved under the strong pressure of sexual selection. Such an evolutionary drive led to the development of sexual dimorphisms in the neural circuitry and neurons that compose this circuitry1. Drosophila melanogaster is an outstanding model organism highly amenable to genetic dissection of complex traits, including mating behavior2. In this organism, the transcription factor gene fruitless (fru) plays a key role in organizing the sexually dimorphic circuitry for mating behavior by specifying sex-specific neuronal structures during development (Sato, 2020).
Among the four promoters identified in the fru gene, the most distal one (the P1 promoter) is dedicated to sex-specific functions ascribed to the male-specific translation of P1-derived mRNAs. As a result of sex-specific splicing of the fru-P1 primary transcript, only male mRNAs have a long ORF encoding male-specific functional fru proteins, called FruM. These proteins are translated as five isoforms, FruAM, FruBM, FruCM, FruDM, and FruEM, which share a common BTB domain in their N-terminus, but which each have a unique C-terminus. The most prevalent isoform is FruBM, which has two zinc-finger motifs in the C-terminus, and forms a complex with chromatin regulators such as the TIF1 homolog Bonus (Bon), histone deacetylase 1 (HDAC1), and heterochromatin protein 1a (HP1a), to bind to more than 100 target sites on the genome for transcriptional regulation of downstream genes. The best-characterized FruBM target is robo1, which encodes a transmembrane protein belonging to the immunoglobulin superfamily with an axon-guidance role in the developing nervous system. FruBM binds to a 42-bp promoter segment of the robo1 gene named the FruBM Response Obligatory Sequence (FROS), which contains a characteristic palindrome sequence of 16 nucleotides (Pal1). Partial deletion of Pal1 in the robo1 gene impairs neural sexual differentiation and mating behavior in male flies, indicating that Pal1 is pivotal for FruBM in order to execute its sexual functions (Sato, 2020).
Many of the fru-expressing neurons are sexually dimorphic, including neurons composing the mAL cluster. The mAL cluster displays sexual dimorphisms in the following four respects: (i) the number of neurons in the cluster (~5 in females vs. ~30 in males); (ii) the absence (females) or presence (males) of a neuronal subset carrying the ipsilateral neurite; (iii) the branched (females) vs. unbranched (males) tip structure of the descending contralateral neurite; and (iv) the focal (females) or expanded (males) terminal arbor distribution of the ascending contralateral neurite. While these four types of sexual dimorphisms of mAL neurons are established in a fru-dependent manner, they are also regulated separately and independently from each other. For example, Hunchback knockdown in male mAL neurons feminizes only the descending contralateral neurite, whereas robo1 knockdown feminizes only the ipsilateral neurite. On the other hand, loss of the cell-death genes, reaper, grim and head involution defective, increases the number of cells composing the mAL cluster from ~5 to ~30 in females, but has no effect in males. Therefore, FruBM likely interacts with different cofactors for transcription regulation, and/or FruBM acts on different targets to establish each of the four sexually dimorphic features of mAL neurons. With the aim of obtaining additional FruBM transcriptional targets for the establishment of neural sexual dimorphisms, this study examined the effects of knocking down the genes harboring a Pal1-homologous motif on mAL neuron structures. teiresias (tei), encoding another immunoglobulin-superfamily member with a transmembrane domain, was identified. Remarkably, knockdown of tei in female mAL neurons completely masculinized all three neurite characteristics, leaving the cell number unaffected. tei knockdown also masculinized fru-expressing sensory neurons and mcALa interneurons in females. It is proposed that the Tei protein functions as a common receptor and interacts with a second receptor, e.g., robo1, which confers the ligand specificity on the heteromeric receptor complex, producing distinct neuronal populations that are diversified in their sex-specific structures (Sato, 2020).
tei, a new transcriptional target of FruBM, was identified based on the in silico search for genes harboring the FruBM-binding consensus sequence and subsequent analysis of gene knockdown effects on sexually dimorphic neurite structures. Tei is a putative transmembrane protein carrying multiple immunoglobulin-like repeats in the extracellular portion. This structure is very much alike that of Robo1, the product of the best-characterized FruBM target gene. Indeed, tei knockdown in female mAL neurons induced the male-specific ipsilateral neurite as did robo1 knockdown, revealing a functional similarity between tei and robo1 in terms of the feminizing effect on this particular structure. Nonetheless, tei was distinctly different from robo1 in that tei is also required for the female-typical shaping of ascending as well as descending contralateral neurites, in which robo1 has no role. Robo1 has been shown to operate as a receptor for the secreted ligand Slit, activating several signal transduction pathways depending on the developmental context in both invertebrates and vertebrates. Promiscuous interactions of Robo1 with other transmembrane receptors in cis have been implicated as a basis for the versatile performances of Robo1 in multiple developmental contexts. The lack of the cytoplasmic portion in Tei might suggest that Tei contributes to the ligand specificity whereas the specificity of intracellular signal transduction following heteromeric receptor activation is determined by Robo1 or another Tei partner that carries the C-terminal cytoplasmic domain. It is therefore plausible that Tei is one of these transmembrane receptors that cooperate with Robo1, and likely initiates together an intracellular biochemical cascade which ultimately inhibits the male-specific ipsilateral neurite from forming in an mAL neuron. Perhaps Tei associates with transmembrane receptors other than Robo1 in specifying the sex-specific structure in the ascending and descending contralateral neurites of an mAL neuron. Systematic knockdown in mAL neurons of genes encoding putative transmembrane receptors with immunoglobulin-like domains other than Robo protein members might lead to the identification of additional Tei partner proteins that are involved in the sex-type specification of the contralateral neurites (Sato, 2020).
Despite its striking effects on neurite structures, tei knockdown had no effect on the sexually dimorphism in the number of mAL neurons. This contrasts with the effects of fru loss-of-function mutation, which reduces the number of cells composing the mAL cluster in males from ~30 to ~5, the cell number typical of the mAL cluster in a wild-type female. Removal of three major cell-death genes, hid, grim and rpr, in female mAL neurons increased the number of cells composing the female mAL cluster up to 29 (the mean cell number was 19). This observation led to the proposition that cell death is the primary cause of the smaller number of cells composing the mAL cluster in females. Moreover, the sex difference in the cell number of fru-expressing neurons has recently been attributed to a difference in proliferation in addition to a difference in cell death: male neuroblasts produce more cells than female neuroblasts, so that males have a larger number of cells even after blocking cell death. These considerations lead to the supposition that FruM proteins separately control the neurite sex-type and the cell number sex-type. In this study it was noted that the Pal1-homologous motif was not found in or around any of the hid, grim, and rpr loci, suggesting they are not direct transcriptional targets of FruM proteins. Alternatively, transcription of these genes might be regulated by a FruM isoform other than FruBM. In fact, the mAL cell number was shown to be affected by the fruB2 (null for FruEM) as well as the fruC1 (null for FruBM) mutation (Sato, 2020).
The 16-bp Pal1 core motif is located within the 42-bp FruBM-binding site, FROS. This motif was first identified in the robo1 gene by a combination of reporter assays, electromobility-shift assays, and CRISPR-Cas9-mediated in vivo mutagenesis in conjunction with phenotypic characterizations of single-cell clones of mAL neurons and mutant-fly behavior. Using different strategies, two other research groups determined the binding consensus sequences for FruAM and FruEM in addition to FruBM. A previous study used the DNA adenosine methyltransferase identification (DamID) method, in which a Fru-fused bacterial methylase methylates the DNA around the genomic region to which the fru moiety of the fusion protein binds. One of the reported FruBM-binding motifs in the DamID-based search revealed a 4/6 match when compared with Pal1, yet robo1 was not obtained as a putative FruBM target in that study. Another study used in vitro screening of oligo-DNAs with random sequences for binding to fru zinc-finger motifs fused to glutathione S-transferase, revealing a binding consensus sequence for each isoform, but none of these had a similarity to Pal1. Of note, CG17716 (tei) has been included, together with another ~1400 genes, in a list of potential FruBM targets having a deduced FruBM-binding sequence. Reliable determination of consensus sequences for the binding of fru isoforms and rigorous identification of fru transcriptional targets are indispensable for obtaining a more complete picture of the molecular mechanisms underlying sex-specific circuit formation (Sato, 2020).
Germ cells in Drosophila melanogaster need intrinsic factors along with somatic signals to activate proper sexual programs. A key factor for male germline sex determination is PHD finger protein 7 (Phf7), a histone reader expressed in the male germline that can trigger sex reversal in female germ cells and is also important for efficient spermatogenesis. This study found that the evolutionarily novel C-terminus in Phf7 is necessary to turn on the complete male program in the early germline of D. melanogaster, suggesting that this domain may have been uniquely acquired to regulate sexual differentiation. Genes were sought that regulated by Phf7 related to sex determination in the embryonic germline by transcriptome profiling of FACS-purified embryonic gonads. One of the genes positively-regulated by Phf7 in the embryonic germline was an HP1 family member, Heterochromatin Protein 1D3 chromoshadow domain (HP1D3csd). This gene is needed for Phf7 to induce male-like development in the female germline, indicating that HP1D3csd is an important factor acting downstream of Phf7 to regulate germline masculinization (Yang, 2021).
This study investigated how Phf7 regulates sex determination in the embryonic germline, and one of the interesting findings is that the unusual C-terminus of Phf7 is necessary for its effects in germline masculinization. The N-terminus of Phf7 is a conserved module comprised of three zinc fingers, of which at least one is functionally essential, and this part of the Phf7 protein evolved from G2E3 (G2/M E3 Ubiquitin Ligase), a protein also made up of three zinc fingers. In contrast, the C-terminus of Phf7 is evolutionarily novel and is not similar to any known domains, suggesting that this domain is undergoing very rapid evolution, a feature not uncommon for factors involved in sex determination (Yang, 2021).
Previously a phylogenetic analysis of Phf7 proteins was conducted across the species tree, and surprisingly it was found that Phf7 in insects and amniotes do not share a common ancestor. Those findings with the latest results indicate that Phf7 in these two animal branches are not orthologous to each other, and that the emergence of the novel C-terminus is likely a unique event that occurred in Drosophila to regulate sexual differentiation in the germline. Recently, mouse Phf7 was demonstrated to be expressed in spermatocytes and can ubiquitinate histones to facilitate histone to protamine exchange. This shows that the expression patterns and functions of D. melanogaster and mouse Phf7 are different, albeit both acting on the male germline. These observations further suggest that the C-terminus of D. melanogaster PHF7 evolved onto an existing module of three zinc fingers, thereby creating new ways to regulating germline sexual development. This is a very interesting example that adds to the collection of diverse mechanisms in sex determination (Yang, 2021).
What does this uncommon C-terminus of PHF7 do? The two most intuitive ideas are that it acts as a transactivation domain like those found in transcription factors, or that it can recruit other effector molecules through protein-protein interactions. The former idea did not hold up when tested in S2 cells. The possibility that the Phf7 C-terminus acts as a bridge between its histone-associating N-terminus and other transcription factors or chromatin factors to alter target gene expression is an appealing one but there is currently no direct data that support this idea (Yang, 2021).
Downstream effectors of Phf7 were sought in the embryonic germline, and it was revealed that HP1D3csd is activated by Phf7 to regulate germline masculinization. Two different genetic tests were perfomed, and while both indicated that Phf7 and HP1D3csd genetically interact, there were some differences in the results. In the Phf7-induced female germline loss assay, both reduction and gain of HP1D3csd expression were found to be exacerbated the Phf7-induced phenotype. In comparison, in the spermatogenesis rescue assay, loss of one HP1D3csd copy hampered rescue whereas HP1D3csd overexpression enhanced spermatogenesis in XX germ cells. The latter experiment is a more direct assay of germline masculinization whereas germline loss can potentially be caused by secondary effects unrelated to sexual development. Therefore, it is thought the results of the spermatogenesis rescue experiments more accurately reflect the relationship between Phf7 and HP1D3csd. In addition to the transcriptome results, HP1D3csd has been identified along with Phf7 to be a part of sex-biased mechanisms in other contexts not limited to the germline. These also support the model that Phf7 and HP1D3csd function synergistically (Yang, 2021).
Phf7 regulates male germline development, and it can associate with the active histone mark methylated H3K47, but it is unclear what Phf7 then does to regulate expression of target genetic loci. H3K9 methylation has also been reported to be important for maintaining sexual differentiation programs in the Drosophila germline. The identification of HP1D3csd as an important downstream factor provides interesting new ideas regarding how the male germline program is initiated and regulated. CSDs have been shown to interact with various chromatin remodelers, thus one appealing model would be that Phf7 can activate or even recruit HP1D3csd to loci important for germline masculinization. This would in turn bring chromatin remodelers to such genes for expression activation and regulation and initiate male-development of the germline. Given the other finding in this study that the C-terminus of Phf7 is an essential part of this process, it would be very interesting to now study which of these factors interact and cooperate with one another (Yang, 2021).
Doublesex (Dsx) and Fruitless (Fru) are the two downstream transcription factors that actuate Drosophila sex determination. While Dsx assists Fru to regulate sex-specific behavior, whether Fru collaborates with Dsx in regulating other aspects of sexual dimorphism remains unknown. One important aspect of sexual dimorphism is found in the gonad stem cell (GSC) niches, where male and female GSCs are regulated to create large numbers of sperm and eggs. This study reports that Fru is expressed male-specifically in the GSC niche and plays important roles in the development and maintenance of these cells. Unlike previously-studied aspects of sex-specific Fru expression, which is regulated by Transformer (Tra)-mediated alternative splicing, this study shows that male-specific expression of fru in the gonad is regulated downstream of dsx, and is independent of tra. fru genetically interacts with dsx to support maintenance of the niche throughout development. Ectopic expression of fru inhibited female niche formation and partially masculinized the ovary. fru is also required autonomously for cyst stem cell maintenance and cyst cell survival. Finally, this study identified a conserved Dsx binding site upstream of fru promoter P4 that regulates fru expression in the niche, indicating that fru is likely a direct target for transcriptional regulation by Dsx. These findings demonstrate that fru acts outside the nervous system to influence sexual dimorphism and reveal a new mechanism for regulating sex-specific expression of fru that is regulated at the transcriptional level by Dsx, rather than by alternative splicing by Tra (Zhou, 2021).
In sexually reproducing animals, the proper production of gametes and successful copulation are equally critical for reproductive success. It is therefore important that both the gonad and the brain know their sexual identity. The Doublesex/Mab-3 Related Transcription Factors act downstream of sex determination and play an evolutionarily conserved role to establish and maintain sexual dimorphism in the gonad. Meanwhile, sexual dimorphism in other tissues such as the brain is controlled, to varying degrees in different animals, through autonomous control by the sex determination and non-autonomous signaling from the gonads. In many invertebrate species, another sex-determination gene fruitless (fru), which encodes multiple BTB-Zinc finger transcription factors, plays a central role in controlling mate choice, courtship behavior and aggression. How sex determination in the gonad and the nervous system are related and coordinated in these species remains unclear (Zhou, 2021).
The founding member of the DMRT family is Drosophila doublesex (dsx). dsx and fru undergo sex-specific alternative mRNA splicing by the sex determination factor Transformer (Tra), together with its co-factor Transformer-2 (Tra-2), to produce transcripts encoding sex-specific protein isoforms. It was once thought that dsx controls sexual dimorphism outside the nervous system while fru regulates sex-specific nervous system development and behavior. But more recent evidence shows that dsx cooperates with fru to specify sex-specific neural circuitry and regulate courtship behaviors. However, whether fru acts along with dsx to control sexual dimorphism outside the nervous system remains unknown (Zhou, 2021).
The fru gene locus contains a complex transcription unit with multiple promoters and alternative splice forms (see Fruitless is expressed male-specifically in the germline stem cell niche and is independent of FruM). Sex-specific regulation of fru was only known to occur through alternative splicing of transcripts produced from the P1 promoter, which produces the FruM isoforms. The downstream promoters (P2-P4) produce Fru isoforms (collectively named FruCom) encoded by transcripts that are common to both sexes and are required for viability in both males and females. fru P1 transcripts have only been detected in the nervous system, suggesting that sex-specific functions of fru are limited to neural tissue. However, FruCom is expressed in several non-neural tissues, including sex-specific cell types of the reproductive system. Further, from a recent genome-wide search for putative Dsx targets, fru was identified as a candidate for transcriptional regulation by Dsx. These data raise the possibility that fru functions cooperatively with dsx to regulate gonad development (Zhou, 2021).
Over the past decades, much effort has been focused on understanding the functions of fru in regulating sex-specific behaviors, yet it remained unclear whether fru plays a role in regulating sexual dimorphism outside the nervous system. The work presented in this study demonstrates that Fru is expressed male-specifically in the gonad stem cell niche, and is required for CySC maintenance, cyst cell survival, and for the maintenance of the hub during larval development. Further, male-specific expression of Fru in the gonad is independent of the previously described mechanism of sex-specific alternative splicing by Tra, and is instead dependent on dsx. fru appears to be a direct target for transcriptional regulation by Dsx. This work provides evidence that fru regulates sex-specific development outside the nervous system and alters traditional thinking about the structure of the Drosophila sex determination pathway (Zhou, 2021).
While it was previously reported that fru is expressed in tissues other than the nervous system, including in the gonad, a function for fru outside the nervous system was previously unknown. This study found that Fru is expressed in the developing and adult testis in the hub, the CySC, and the early developing cyst cells. Importantly, it was found that fru is important for the proper function of these cells (Zhou, 2021).
Fru is not expressed at the time of hub formation during embryogenesis, but expression is initiated during the L2/L3 larval stage. This correlates with a time period when the hub must be maintained and resist transforming into female niche structures: in dsx mutants, all gonads in XX and XY animals develop hubs, but in half of each, hubs transform into terminal filament cells and cap cells. fru is not required for initial hub formation, consistent with it not being expressed at that time. fru is also not, by itself, required for hub maintenance under the conditions that were possible to assay (prior to the pupal lethality of fru null mutant animals). However, under conditions where hub maintenance is compromised by loss of dsx function, fru clearly plays a role in influencing whether a gonad will retain a hub, or transform into TF. Fru expression in dsx mutant gonads correlates with whether they formed male or female niche structures, and removing even a single allele of fru is sufficient to induce more hubs to transform into TFs. Finally, ectopic expression of Fru in females is sufficient to inhibit TF formation and partially masculinize the gonad, but does not induce hub formation. Thus, it is proposed that fru is one factor acting downstream of dsx in the maintenance of the male gonad stem cell niche, but that it acts in combination with other factors that also regulate this process (Zhou, 2021).
This study also demonstrated that fru is required for CySC maintenance and for the survival of differentiating cyst cells. Loss of fru from the CySC lineage led to rapid loss of these CySCs from the testis niche. Since precocious differentiation of CySCs or an increase in their apoptosis was not observed, these mechanisms do not appear to contribute to CySC loss. One possibility is that fru is needed for CySCs to have normal expression of adhesion proteins and compete with other stem cells for niche occupancy. It has been shown that fru regulates the Slit-robo pathway and that robo1 is a direct target of fru in the CNS. Interestingly, the Slit-Robo pathway also functions in the CySCs to modulate E-cadherin levels and control the ability of CySCs to compete for occupancy in the niche. Therefore, fru may use similar mechanisms to maintain CySC attachment to the hub. fru also influences survival in the differentiating cyst cells, as an increase in cell death was observed in these cells in fru mutants. Several reports have demonstrated that fru represses programmed cell death in the nervous system. It was further indicated that the cell death gene reaper is a putative target of Fru. Thus, fru may play a role in repressing the apoptosis of cyst cells (Zhou, 2021).
In summary, fru function is clearly important for male niche maintenance and the function of the CySCs and their differentiating progeny. This provides clear evidence that fru regulates sex-specific development in tissues other than the nervous system. Whether additional tissues are also regulated by fru remains to be determined (Zhou, 2021).
Previously, it was thought that the only mechanism by which sex-specific functions of fru were regulated was through Tra-dependent alternative splicing of the P1 transcripts. fru null alleles are lethal in both sexes and Fru proteins derived from non-P1 promoters were thought to be sex-nonspecific and not to contribute to sex determination. Thus, fru and dsx were considered as parallel branches of the sex determination pathway, each independently regulated by Tra. This study demonstrates that fru can also be regulated in a manner independent of tra and dependent on dsx, and provides evidence that fru is a direct target for transcriptional regulation by Dsx (see Proposed model of the Drosophila sex determination pathway. First, Fru expression in the testis is independent of the P1 transcript that is regulated by Tra. A P1 Gal4 reporter is not expressed in the testis and a mutation that prevents FruM expression from P1 does not affect Fru immunoreactivity in the testis. Second, in animals that simultaneously express the female form of tra (Tra on) and the male form of Dsx [XX; dsxD/Df(3R)dsx3], Fru is expressed in the male mode in the testis, demonstrating that it is regulated by dsx and not tra. Finally, an evolutionarily conserved Dsx consensus binding site upstream of the P4 promoter is required for proper expression levels of a fru P4 reporter in the testis. Together, these data demonstrate a novel mode for fru regulation by the sex determination pathway, where sex-specific expression of fru is regulated by dsx. It also means that the large number of fru transcripts that do not arise from the P1 promoter can be expressed in a sex-specific manner to contribute to sexual dimorphism (Zhou, 2021).
The male and female forms of Dsx contain the same DNA binding domain and can regulate the same target genes, but often have opposite effects on gene expression. Prior to this study, the documented Dsx targets (Yolk proteins 1 and 2, bric-a-brac and desatF), along with other proposed targets, were all expressed at higher levels in females than males. Thus, for these targets, DsxF acts as an activator and DsxM acts as a repressor (or DsxM has no role). Interestingly, fru is the first identified Dsx target that is expressed in a male-biased manner. Thus, for direct regulation of fru, DsxM would activate expression while DsxF represses. Mechanistically for Dsx, this implies that the male and female isoforms are not dedicated repressors and activators, respectively, but may be able to switch their mode of regulation in a tissue-specific or target-specific manner. Mouse DMRT1 has also been shown to regulate gene expression both as transcriptional activator and repressor. Thus, it is quite possible that bifunctional transcriptional regulation is a conserved characteristic of DMRTs (Zhou, 2021).
It is possible that dsx regulation of fru occurs in the nervous system as well, where it co-exists with direct regulation of fru alternative splicing by Tra. It was originally thought that alternative splicing of the fru P1 transcript by tra was essential for male courtship behavior. However, more recently it was found that these animals could exhibit male courtship behavior if they were simply stimulated by other flies prior to testing. Interestingly, the courtship behavior exhibited by these males was dependent on dsx. It is proposed that fru might still be essential for male courtship in these fru P1-mutants, but that sex-specific fru expression is dependent on transcriptional regulation of other fru promoters by Dsx (Zhou, 2021).
If sex-specific fru function can be regulated both through alternative splicing by Tra and through transcriptional regulation by Dsx, it raises the question of what is the relationship between these two modes of regulation? It is proposed that regulation of fru by Dsx is the more ancient version of the sex determination pathway and that additional regulation of fru by Tra evolved subsequently, through the acquisition of regulatory RNA elements in the fru P1 transcript. This model is supported by studies of fru gene structures in distantly related Dipteran species, and species of other insect orders, that illustrate the considerable variability in the organization of sequences controlling fru splicing. Further, in some insects, no evidence for alternative splicing of fru has been found, yet fru still plays an important role in males to control courtship behaviors. Finally, in the Hawaiian picture-winged group of subgenus Drosophila, the fru orthologues lack the P1 promoter, and non-P1 fru transcripts exhibit male-specific expression, similar to what is proposed for non-P1 fru transcripts in D. melanogaster. Thus, it appears that regulation of fru by dsx may be the evolutionarily more ancient mechanism for sex-specific control of fru, while Tra-dependent splicing of P1 transcripts is a more recent adaptation. More broadly, tra is not conserved in the sex determination pathway in the majority of animal groups, while homologs of Dsx, the DMRTs, are virtually universal in animal sex determination. Thus, if Fru orthologs are involved in the creation of sexual dimorphism in the body or the brain in other animals, they cannot be regulated by Tra but may be regulated by DMRTs (Zhou, 2021).
Light exposure impacts several aspects of Drosophila development including the establishment of circadian rhythms, neuroendocrine regulation, life-history traits, etc. Introduction of artificial lights in the environment has caused almost all animals to develop ecological and physiological adaptations. White light which comprises different lights of differing wavelengths shortens the lifespan in fruit flies Drosophila melanogaster. The wavelength-specific effects of white light on Drosophila development remains poorly understood. This study shows that different wavelengths of white light differentially modulate Drosophila development in all its concomitant stages when maintained in a 12-h light: 12-h dark photoperiod. It was observed that exposure to different monochromatic lights significantly alters larval behaviours such as feeding rate and phototaxis that influence pre-adult development. Larvae grown under shorter wavelengths of light experienced an altered feedingrate. Similarly, larvae were found to avoid shorter wavelengths of light but were highly attracted to the longer wavelengths of light. Most of the developmental processes were greatly accelerated under the green light regime while in other light regimes, the effects were highly varied. Interestingly, pre-adult survivorship remained unaltered across all light regimes but light exposure was found to show its impact on sex determination. This study for the first time reveals how different wavelengths of white light modulate Drosophila development which in the future might help in developing non-invasive therapies and effective pest measures (Ramakrishnan, 2022).
The evolution of sex chromosomes has resulted in numerous species in which females inherit two X chromosomes but males have a single X, thus requiring dosage compensation. MSL (Male-specific lethal) complex increases transcription on the single X chromosome of Drosophila males to equalize expression of X-linked genes between the sexes. The biochemical mechanisms used for dosage compensation must function over a wide dynamic range of transcription levels and differential expression patterns. It has been proposed that the MSL complex regulates transcriptional elongation to control dosage compensation, a model subsequently supported by mapping of the MSL complex and MSL-dependent histone 4 lysine 16 acetylation to the bodies of X-linked genes in males, with a bias towards 3' ends. However, experimental analysis of MSL function at the mechanistic level has been challenging owing to the small magnitude of the chromosome-wide effect and the lack of an in vitro system for biochemical analysis. This study used global run-on sequencing (GRO-seq) to examine the specific effect of the MSL complex on RNA Polymerase II (RNAP II) on a genome-wide level. Results indicate that the MSL complex enhances transcription by facilitating the progression of RNAP II across the bodies of active X-linked genes. Improving transcriptional output downstream of typical gene-specific controls may explain how dosage compensation can be imposed on the diverse set of genes along an entire chromosome (Larschan, 2011).
In addition to increasing the transcription of X-linked genes for dosage compensation, MSL complex also positively regulates the roX noncoding RNA components of the complex, to promote their male-specificity. roX1 expression is low in the SL2 cell line, but GRO-seq data indicate that active transcription of roX2 is highly dependent on MSL2 as predicted. Interestingly, there is a strong GRO-seq peak at the 3′ roX2 DHS which contains sequences important for targeting MSL complex to the X chromosome. Sites of roX gene transcription are thought to be critical for MSL complex assembly. Therefore, it is possible that paused RNAP II at the roX2 DHS could promote an open chromatin structure that facilitates MSL complex targeting or incorporation of noncoding roX2 RNA into the complex (Larschan, 2011).
In summary, it is hypothesized that MSL complex functions on the male X chromosome to promote progression and processivity of RNAP II through the nucleosomal template. Improving transcriptional output downstream of typical gene-specific regulation makes biological sense when compensating the diverse set of genes found along an entire chromosome (Larschan, 2011).
Dosage compensation in Drosophila is mediated by the MSL complex, which increases male X-linked gene expression approximately 2-fold. The MSL complex preferentially binds the bodies of active genes on the male X, depositing H4K16ac with a 3' bias. Two models have been proposed for the influence of the MSL complex on transcription: one based on promoter recruitment of RNA polymerase II (Pol II), and a second featuring enhanced transcriptional elongation. This study utilize nascent RNA sequencing to document dosage compensation during transcriptional elongation. Also, comparison was made of X and autosomes from published data on paused and elongating polymerase in order to assess the role of Pol II recruitment. The results support a model for differentially regulated elongation, starting with release from 5' pausing and increasing through X-linked gene bodies. The results highlight facilitated transcriptional elongation as a key mechanism for the coordinated regulation of a diverse set of genes (Ferrari, 2013).
This study has systematically dissected the mechanism
of DC during distinct steps of transcription. Multiple high-resolution,
genome-wide approaches converge on the following model: paused Pol II is not augmented in general on the male
X, but Pol II release from pausing ('jump-start' in the model)
appears to be a key rate-limiting step that is facilitated by X-specific
enrichment of H4K16ac in gene bodies. The increasing MSL
and H4K16ac levels over the bodies of genes further reduce
steric hindrance, leading to a 'gain' of Pol II density. Currently,
it cannot be determined whether this gain is the result of (1)
increased processivity (reduced termination) or (2) positive feedback
to 5' Pol II, to further increase pausing release. In either
case, it is believed that facilitated elongation through an acetylated
chromatin template enables coordinate control of X-linked
genes with widely differing mechanisms of individual, gene-specific
regulation (Ferrari, 2013).
In Drosophila, two chromosome-wide compensatory systems have been characterized: the dosage compensation system that acts on the male X chromosome and the chromosome-specific regulation of genes located on the heterochromatic fourth chromosome. Dosage compensation in Drosophila is accomplished by hypertranscription of the single male X chromosome mediated by the male-specific lethal (MSL) complex. The mechanism of this compensation is suggested to involve enhanced transcriptional elongation mediated by the MSL complex, while the mechanism of compensation mediated by the Painting of fourth (POF) protein on the fourth chromosome has remained elusive. This study shows that POF binds to nascent RNA, and this binding is associated with increased transcription output from chromosome 4. Genes located in heterochromatic regions spend less time in transition from the site of transcription to the nuclear envelope. These results provide useful insights into the means by which genes in heterochromatic regions can overcome the repressive influence of their hostile environment (Johansson, 2012).
Aneuploidy of entire chromosomes and chromosome segments is an important evolutionary driving force that increases variation but is accompanied by problems associated with changes in gene dosage and genomic instability. The evolution of buffering systems that compensate for dosage differences must therefore allow for a balance between allowing genomic variability and avoiding genomic instability. Buffering systems of this kind have been described in Drosophila; in haploid conditions, they cause the transcription output to increase by a factor of approximately 1.4. However, the evolution of heteromorphic sex chromosomes, such as the X and Y chromosome pair in flies and mammals, is accompanied by an expression problem that requires more extensive compensation. Since most genes on the X chromosome should be expressed at the same levels in males and females, dosage compensation mechanisms coevolve as the X-Y chromosome pair is formed. Notably, while the ancient homology between the mammalian X and Y is clear, the evolutionary origin of the Drosophila Y is more complicated. The evolution of dosage compensation mechanisms is attributable to evolutionary pressures that act at all levels of expression to compensate for the losses of functional gene copies. Two dosage compensation systems have been studied in Drosophila: the male-specific lethal (MSL) complex, which targets and upregulates the male X chromosome, and the painting of fourth (POF) protein, which stimulates the expression of the fourth chromosome in Drosophila melanogaster but is believed to have originated from a dosage-compensating mechanism. The MSL complex and POF coexist; they are on different chromosomes in, e.g., D. melanogaster, but are colocalized on the same chromosome in, e.g., Drosophila ananassae. Their coexistence suggests that they probably act on different levels of gene regulation (Johansson, 2012).
The MSL complex is a ribonucleoprotein complex consisting of five male-specific lethal proteins (MSL1, MSL2, MSL3, MLE, and MOF) and two noncoding RNAs, roX1 and roX2. Because expression of MSL2 is sex restricted, the complex is formed only in males and specifically targets the male X chromosome. A model of its action has been proposed: MOF (a histone acetyltransferase) acetylates H4K16, and this modification leads to decompaction of the chromatin and hypertranscription of the male X chromosome genes. The prevailing idea is that the MSL complex stimulates transcriptional elongation. This idea is supported by genome-wide mappings showing that expression of the MSL complex and the associated H4K16 acetylation are both enhanced within gene bodies with a bias to their 3' end. A recent study confirmed that the MSL complex enhances transcription by facilitating the progression of RNP2 across the active X chromosomal genes (Johansson, 2012).
Less is known about the regulatory level at which POF acts. POF is a 55-kDa protein containing an RNA recognition motif (RRM). Like the MSL complex, POF binds within the bodies of expressed genes. However, in D. melanogaster, POF specifically targets the fourth chromosome in both males and females. The targeting of POF to the fourth chromosome is associated with a chromosome-specific increase in transcript levels that primarily affects differentially expressed genes. Flies can survive without POF or missing one copy of the fourth. However, haplo-fourth animals die if they also lack POF. Importantly, the expression of nonubiquitously expressed genes on the fourth chromosome has been shown to be compensated by POF in haplo-4 flies; suppressing or eliminating this compensation causes haplo-fourth lethality (Johansson, 2012).
The fourth chromosome of D. melanogaster has several unique characteristics. It is the smallest chromosome in the Drosophila genome, with an approximate size of 5 Mb. Of these 5 Mb, 3 to 4 Mb consists exclusively of simple AT-rich satellite repeats and does not contain any known genes. The sequenced part of chromosome 4 is only 1.3 Mb and represents the banded, polytenized, and gene-rich portion corresponding to cytological sections 101E to 102F. In principle, the entire fourth chromosome can be considered heterochromatic; more specifically, it consists of the HP1 enriched 'green chromatin' as defined by van Steensel and coworkers. This means that the fourth chromosome is enriched in the heterochromatin protein HP1 and in specific histone modification markers of heterochromatin, e.g., methylated H3K9. In keeping with its heterochromatic nature, the polytenized part of chromosome 4 contains large blocks of repeated sequences and transposable elements that are interspersed with the genes. Transgenes inserted on the fourth chromosome often show partially silenced, variegated expression. In fact, the structure and sequence composition of the fourth chromosome, with its scattered repetitive elements, is more reminiscent of the organization of mammalian chromosomes than that of the other D. melanogaster autosomes. It appears as though the genes located on the fourth chromosome have adapted to function in this repressive milieu (Johansson, 2012).
To study the fundamental compensatory processes acting on the fourth chromosome, RNA immunoprecipitation (RIP) experiments followed by tiling array analysis (RIP-chip) and transcript profiling experiments were performed. This study shows that POF associates with the newly transcribed RNA produced from the fourth chromosome. The data indicate that POF binds to the spliced form of the transcript and that this binding is associated with an increase in the amount of chromosome 4 transcripts. It was also shown that transcripts encoded by the fourth chromosome or pericentromeric heterochromatin have a shorter transition time from the site of transcription to the nuclear envelope (Johansson, 2012).
In Drosophila, two chromosome-wide compensatory systems have been characterized: the MSL complex, which targets and stimulates the expression of the X chromosome in males, and POF, which targets and stimulates the expression of the fourth chromosome. It has been hypothesized that the MSL complex stimulates expression by facilitating transcriptional elongation, and this hypothesis was recently confirmed experimentally. This study has used RNA immunoprecipitation and transcriptome profiling techniques to further clarify the mechanism by which POF stimulates gene expression (Johansson, 2012).
Previous studies have shown that POF binds to active genes on the fourth chromosome. Since previously reported ChIP-chip results were obtained using cross-linked extracts, they could not be used to determine whether POF associates directly with the chromatin or binds via interactions with other components and was frozen in place by the cross-linking. In the work reported in this study, a relationship was observed between the binding of the POF protein to the chromosome and that of the RNA polymerase. This connection with transcription, together with the fact that POF possesses an RNA binding domain, suggested that POF binds to RNA rather than directly to chromatin. To test this hypothesis, a genome-wide POF RIP-chip experiment was performed. The POF-RIPs verified the association of POF with chromosome 4 transcripts (Johansson, 2012).
Two components of the MSL complex (MSL2 and MOF) were also investigated, both as controls and to determine whether the MSL complex also binds to transcripts from the chromosome it regulates, i.e., the male X chromosome. In contrast to the observed association of POF with transcripts from the fourth chromosome, no unambiguous evidence was found to support any binding of MSL2 or MOF to X-linked transcripts. However, at this point, it cannot be excluded that other components of the MSL complex, especially the more loosely bound MLE, may have a general affinity for transcripts of the X chromosome. A slight reduction was also noticed in the number of transcripts from the fourth chromosome in both the MSL2-RIPs and the MOF-RIPs, suggesting that the general RNA affinity of the MSL complex is less pronounced for the fourth chromosome than for the other chromosomes. This relative reduction in the number of chromosome 4 transcripts associated with the MSL complex might reflect the binding of POF to these transcripts, which could block their association with other RNA binding proteins (Johansson, 2012).
The POF-RIP results, together with the previously reported strong link between POF and the fourth chromosome, suggested that POF binds to nascent RNAs, while these RNAs are still bound to the RNP2. The most parsimonious explanation of the high similarity between the ChIP-chip and RIP-chip profiles for POF is that POF associates with nascent RNA and the ChIP profile is indicative of chromatin linked via RNP2. However, at this point it cannot be excluded that POF in addition to binding nascent RNAs also associates with fully processed mature mRNAs en route to the cytoplasm. It also remains possible that POF, in addition to binding nascent RNA, also associates with chromatin via interactions with other proteins (for instance, HP1). No evidence of physical association between POF and HP1 has so far been reported, but POF and Setdb1 (the HKMT responsible for H3K9me on the fourth chromosome) have been shown to interact in vitro. Thus, POF may be an element of an adaptor system linking histone marks to nascent RNA via HP1 and Setdb1, in a fashion similar to MRG15 and PTB. The fact that POF binding to chromatin is RNase resistant may be explained by a stabilization of POF via interaction with a chromatin-associated factor like HP1 or Setdb1. The RNase resistance may also be caused by inaccessibility, i.e., the nascent RNA associated with POF is not accessible to the RNase (Johansson, 2012).
POF was observed to bind to RNA from other chromosomes, indicating that it possesses a general affinity for RNA. This association with transcripts other than those from the fourth chromosome is more pronounced in the native samples, suggesting that it occurs during sample preparation as an equilibrium reaction rather than accurately reflecting in vivo associations. It is speculated that during the preparation of the native samples, some POF molecules are released from their normal target sites and become free to associate with any transcript in the nucleoplasm. In contrast, in the cross-linked samples, the in vivo POF binding is 'frozen' before any sample treatment steps are performed. Consequently, POF will only be observed to bind to chromosome 4 transcripts, and the apparent enrichment of RNAs encoded from other chromosomes is lost (Johansson, 2012).
Splicing is commonly regarded as a process that takes place after transcription. However, cotranscriptional splicing was visualized in Drosophila more than 25 years ago. More recent studies have revealed that cotranscriptional splicing is more common than was previously believed and also that splicing can begin as a cotranscriptional event and continue posttranscriptionally . Therefore, the strong exon bias reported in this study is compatible with POF binding to nascent RNAs. Cotranscriptional splicing is believed to depend on cooperation between exon recognition and the speed of transcription by RNP2. Further, it has been shown that the density of nucleosomes is higher within exons; they may thus function as 'speed bumps' to slow down the RNP2 elongation rate. Since POF is connected to nascent RNAs, this reduction in the speed of transcription over exons would explain the exon bias observed at the chromatin level in ChIP-chip experiments (Johansson, 2012).
Given that POF presumably binds to nascent RNA via its RRM1 domain, it is interesting to consider hypothetical mechanisms by which POF might regulate the expression of genes on chromosome 4. The binding of POF to nascent RNA may directly or indirectly (i.e., via chromatin structure modifications) stimulate transcription. There are several examples of interactions between chromatin-associated proteins recognizing histone modification marks and proteins that bind nascent RNA (Johansson, 2012).
To explore the potential difference in engaged RNP2 distribution on chromosome 4 compared to other autosomes, the previously published GRO-seq data (that maps the position, amount, and orientation of transcriptionally engaged polymerases genome wide) for Drosophila S2 cells was used. That study shows that the male X chromosome has a higher elongation density index (EdI) than the autosomes, which is interpreted as an enhanced transcription elongation. In contrast, comparing the fourth chromosome to all other autosomes, a significantly decreased EdI was found, consistent with a less efficient transcription elongation. The fourth chromosome also shows a decreased pausing index (PI). This is, on the other hand, in line with an increased transcription output from chromosome 4 genes. It is tempting to speculate that the heterochromatic nature of the fourth chromosome, with HP1 enriched over gene bodies of active genes, causes the decreased EdI. Considering that the fourth chromosome is expressed at levels equal to (or slightly higher than) those of the other chromosomes, this elongation disadvantage may be counteracted by a decreased RNP2 pausing. Since POF is bound to in principle all active chromosome 4 genes, it remains elusive whether POF is connected to the observed decrease in chromosome 4 PI (Johansson, 2012).
It is also possible that the binding of POF to nascent RNA has posttranscriptional effects. There are at least three possible posttranscriptional scenarios we must consider: splicing, protection, and transport. If splicing were the main function of POF targeting, we would expect a difference in the transcriptome profiles between Pof mutants and the wild type. However, the transcriptome profiles of Pof mutants are very similar to those of the wild type, and there is no evidence of an increased rate of incorrect splicing or more frequent use of introns in the Pof mutant. The only striking difference between the transcriptome profile of the Pof mutant and that of the wild type is the reduction in the amount of processed transcripts from chromosome 4. The same reduction is observed whether we look at the 5' end, 3' end, or middle part of the genes and is thus less consistent with the hypothesis that chromosome 4 transcripts are more prone to degradation in Pof mutants. This demonstrates that POF has a positive effect on the amount of chromosome 4 transcripts which is not caused by improved splicing efficiency (Johansson, 2012).
Analysis of the ratio of input RNA levels from WCFA (whole cells) to input RNA levels from FA (nuclei) revealed that the relative amounts of chromosome 4 transcripts and transcripts from genes in the pericentromeric region are higher in the cytoplasm than in the nucleoplasm, in relation to transcripts from the other chromosomes. Notably, although export rates per se have not been measured, the results are consistent with pericentromeric and chromosome 4 transcripts being more efficiently exported. Chromatin is highly organized within the nucleus: euchromatic blocks are preferentially located in the center, while heterochromatic regions, such as the fourth chromosome and pericentromeric heterochromatin, tend to localize closer to the nuclear rim. Whether the nuclear periphery is a repressive or permissive environment for gene expression has been debated. The transition time for the export of transcribed mRNAs from the site of synthesis to the nuclear pore would be minimized for chromosomal regions close to the nuclear pore. The gene-gating hypothesis postulates that nucleoporins associate with active genes and facilitate the export of the corresponding mRNAs. Components of the nuclear pore complex (nucleoporins) have been reported to interact with transcriptionally active genes. It has been shown in a mammalian system that transport of an mRNA from the site of transcription to the nuclear pore occurs within a time frame of 5 to 40 min. In the same study, no pileup of mRNAs at the nuclear pore was found, and export through the pore was rapid (0.5 s). Thus, a closer proximity to the nuclear pore may increase transcription output, especially in rapidly dividing cells, since reducing the transition time would allow a more rapid initiation of protein synthesis after cell division. It was observed that the whole cell/nuclei ratio for transcripts produced from the fourth chromosome and the pericentromeric heterochromatin was increased compared to the transcript ratio of the entire genome. It should be stressed that although these genes are located in seemingly repressive environments, both the number of expressed genes and gene expression are comparable to euchromatic chromosome regions. It may be that genes located in these heterochromatic regions benefit from their relative proximity to the nuclear pore, which would facilitate the export of transcribed mRNA. This may in fact be one reason why genes located in pericentric heterochromatin, such as light and rolled, are repressed by rearrangements that move them into euchromatic surroundings (Johansson, 2012).
It is tempting to speculate that the evolution of more efficient logistics for the export of transcripts from the fourth chromosome was driven by the need to facilitate the expression of its genes. This study has also shown that the quantity of chromosome 4 transcripts is reduced in Pof mutants, and the data indicate that this decrease is not caused by splicing defects or increased degradation. It is not yet known whether the binding of POF to nascent RNAs increases the efficiency of transcription or whether it facilitates their efficient export. However, it should be stressed that transcription levels are probably influenced by a number of stimulatory and repressive influences and that during the course of evolution these factors become increasingly interdependent (Johansson, 2012).
Transposable elements (TEs) may contribute to evolutionary innovations through the rewiring of networks by supplying ready-to-use cis regulatory elements. Genes on the Drosophila X chromosome are coordinately regulated by the male specific lethal (MSL) complex to achieve dosage compensation in males. This study shows that the acquisition of dozens of MSL binding sites on evolutionarily new X chromosomes, in Drosophila miranda, was facilitated by the independent co-option of a mutant helitron TE that attracts the MSL complex (TE domestication). The recently formed neo-X recruits helitrons that provide dozens of functional, but suboptimal, MSL binding sites, whereas the older XR chromosome has ceased acquisition and appears to have fine-tuned the binding affinities of more ancient elements for the MSL complex. Thus, TE-mediated rewiring of regulatory networks through domestication and amplification may be followed by fine-tuning of the cis-regulatory element supplied by the TE and erosion of nonfunctional regions (Ellison, 2013).
Active transposable elements (TEs) impose a substantial mutational burden on the host genome. However, there is growing evidence implicating TEs as drivers of key evolutionary innovations by creating or rewiring regulatory networks. Many TEs harbor a variety of regulatory motifs, and TE amplification may allow for the rapid accumulation of a specific motif throughout the genome, thus recruiting multiple genes into a single regulatory network (Ellison, 2013).
In Drosophila miranda, multiple sex chromosome/autosome fusions have created a series of X chromosomes of differing ages. The ancestral X chromosome, XL, is homologous to the D. melanogaster X and is at least 60 million years old. Chromosome XR became a sex chromosome ~15 million years ago and is shared among members of the affinis and pseudoobscura subgroups, whereas the neo-X chromosome is specific to D. miranda and originated only 1 million years ago. The male specific lethal (MSL) complex coordinates gene expression on the Drosophila male X to achieve dosage compensation. This complex is recruited to the X chromosome in males to high-affinity chromatin entry sites (CES) containing a conserved GA-rich sequence motif, roughly 21-base pair (bp) in length, termed the MSL recognition element (MRE). Once bound, the MSL-complex spreads from the CES in cis to actively transcribed genes, where it catalyzes the deposition of the activating histone modification H4K16ac, which ultimately results in a chromosome-wide twofold increase in gene expression levels. D. miranda males show MSL binding specific to the X chromosomes, associated with full dosage compensation of chromosomes XL and XR. In contrast, the neo-X shows incomplete dosage compensation (Ellison, 2013).
The evolution of dosage compensation on XR and the neo-X involved co-option of the MSL machinery (Marin, 1996) and the creation of CES capable of recruiting this machinery, via MRE sequence motifs at a few hundred locations along the two X chromosomes. This study used chromatin immunoprecipitation sequencing (ChIP-seq) profiling of MSL binding to conservatively define 132 CES on chromosome XL, 215 on XR, and 68 on the neo-X, and a more realistic estimate identifies 219 CES on XL, 383 on XR, and 175 on the neo-X; these two groups are referred to as 'strict' versus 'broad' set of CES. The CES on XR and the neo-X likely arose within the past 15 and 1 million years, respectively, after these chromosomes became X-linked in an ancestor of D. miranda (Ellison, 2013).
Comparison of the genomic regions at strict neo-X CES sequences to their homologous regions in D. pseudoobscura, which are not X-linked and do not recruit the MSL complex, identified the mutational paths responsible for the formation of a MRE at 41 CES on the neo-X. In half of these sites, point mutations and short indels at prebinding sites created a stronger MRE. For the remaining half, however, the new MREs appeared to have been gained via a relatively large (~1 kb), D. miranda-specific insertion. Sanger resequencing and manual curation of the genome assembly at these sites allowed it to be determined that these insertions are derived from a transposable element (homologous to the ISY element) that is highly abundant in the genome of D. miranda and its relatives (>1000 copies in D. miranda and D. pseudoobscura). The ISY element (~1150 bp) is a nonautonomous helitron, a class of DNA-transposable elements that replicate through a rolling-circle mechanism. All 21 elements found at strict CES on the neo-X share a 10-bp deletion relative to the consensus ISY element, and the ISY sequence containing this deletion is referred to as ISX. ISX is also found at 24 of broad CES and is present at 43% of strict CES and 30% of broad CES on the neo-X. This 10-bp deletion creates a sequence motif more similar to the consensus MRE motif inferred from XL relative to the consensus ISY sequence and thus might create a strong recruitment signal for the MSL-complex. The ISX element - but not ISY - is specific to D. miranda and highly enriched on the neo-X relative to other chromosomes and strongly bound by the MSL complex in vivo. Additionally, the sequence similarity among ISX elements found at CES on the neo-X is consistent with their recent acquisition on the neo-X, after the formation of the neo-sex chromosomes. Together, these results suggest that within the past 1 million years, the D. miranda lineage was invaded by a domesticated helitron that recruits hundreds of genes into the MSL regulatory network on the neo-X. This process involved the formation of a high-affinity MRE sequence motif via a 10-bp deletion, followed by amplification and fixation of this element at dozens of sites along the neo-X chromosome(Ellison, 2013).
A transgenic assay was used in D. melanogaster to functionally verify that the ISX element attracts the MSL complex and functions as a CES. The construct was targeted to the previously characterized autosomal landing site 37B7 in D. melanogaster. Immunostaining of male polytene chromosomes shows that the ISX element can recruit the MSL complex of D. melanogaster, but no staining was detected with the ISY elemen. A higher affinity of the MSL complex to ISX versus ISY was also confirmed by means of ChIP-quantitative polymerase chain reaction. Mutagenesis assays were used to convert this ISX element into ISY by inserting the 10-bp sequence (ISX ->: ISY), and the 10-bp fragment was deleted from the ISY element to create ISX (ISY ->: ISX). Immunostaining confirmed that the ISX ->: ISY construct could no longer recruit the MSL-complex to an autosomal location, whereas the ISY ->: ISX transgene was now able to attract MSL to an autosomal landing site in D. melanogaster. Thus, the ISX element alone is able and sufficient to attract the MSL complex, and the 10-bp deletion creates a functional MSL recruitment site. This experimentally confirms that the amplification of this TE along the neo-X chromosome may have resulted in the rapid wiring of neo-X-linked genes into the dosage compensation network. Dosage compensation of neo-X genes is advantageous because ~40% of homologous neo-Y genes are pseudogenized; however, because of its ability to recruit the MSL complex and induce dosage compensation, the ISX element should be selected against from autosomal locations. Indeed, out of a total of 82 copies of the ISX element, only two exist on an autosome, within repeat-rich and supposedly silenced regions on the dot chromosome (Ellison, 2013).
In the ancestor of the affinis and pseudoobscura subgroups (~15 million years ago), Muller element D became incorporated into the dosage compensation network after it fused to the ancestral X to form chromosome XR. All CES sequences on XR were compared to determine whether they were enriched for sequence elements besides the MRE motif that would be indicative of a TE burst. Three repeat elements were present in ~22% of strict (and in 14.4% of broad) XR CES sequences, but not in the homologous regions from D. subobscura, where this chromosome is an autosome. Furthermore, these elements were all determined to be conserved fragments from a single TE (hereafter referred to as ISXR), which is derived from the same helitron family as the ISY/ISX elements. Individual ISXR copies are less similar to each other than the ISX elements, and sequence divergence among the different copies of this TE is consistent with a burst of transposition activity coinciding with the formation of chromosome XR. Additionally, ISXR is enriched on chromosome XR, and similar to ISX/ISY, its autosomal homologs show less sequence similarity to the MRE consensus motif and cannot recruit the MSL-complex in vivo. ISXR contains a ~350-bp region that is not present in any of the ISY or ISX elements, and this region specific to ISXR contains an additional MRE motif in close proximity to the MRE whose location is conserved between the ISX and ISXR elements. In addition, although the location of the 3' ISXR MRE is conserved with ISX, there is no evidence of the 10-bp deletion seen in ISX. The presence of this particular sequence region suggests that although ISX and ISXR evolved from a similar helitron progenitor TE, they represent independent TE domestications and chromosomal expansions at different time points. Consistent with the more ancient expansion of ISXR, nonfunctional parts of the TE are severely eroded (Ellison, 2013).
Similarity-based clustering of the MRE consensus motifs from each helitron subtype reveal that both ISXR MRE motifs are more similar to the canonical XL MRE motif, compared with the ISX MRE motif. This suggests that MSL binding motifs supplied by ISX may be suboptimal, whereas ISXR binding affinity is optimized. A large number of substitutions observed at MRE motifs among ISXR copies across the genome and elevated rate of evolution at homologous ISXR MRE sites relative to XL MREs across species suggest that the ISXR element initially may have also harbored a suboptimal MRE motif. Over time, mutation and selection may have fine-tuned the nucleotide composition at ISXR independently across elements and species, to maximize MSL recruitment by increasing their similarity to the canonical XL MRE motif. In agreement with this observation, the TE-derived XR CES show a higher affinity for MSL complex in vivo as compared with that of those on the neo-X (Ellison, 2013).
The recently formed sex chromosomes of D. miranda provide insights into the role of TEs in rewiring regulatory networks. The evolutionary pressure driving the acquisition of dosage compensation as well as the molecular mechanism of MSL function and targeting provide clear expectations of which genes should be recruited into the dosage compensation network, as well as when and how. Additionally, the comparison of XR and the neo-X allows a study of the dynamic process of TE-mediated wiring of chromosomal segments into the dosage-compensation network at two different evolutionary stages: both the initial incorporation of the neo-X chromosome by amplification of a domesticated TE and possible subsequent fine-tuning of the regulatory element supplied by the TE on XR, together with the erosion of TE sequence not required for MSL-binding. The data support a three-step model for TE-mediated rewiring of regulatory networks (domestication, amplification, and potential fine-tuning) followed by erosion of nonfunctional parts of the transposon. Eventually, the footprints left behind by TE-mediated rewiring will completely vanish, and many ancient bursts of domesticated TEs that rewired regulatory networks are likely to go undetected. Indeed, no TE relics within the CES of chromosome XL were detected that acquired MSL-mediated dosage compensation over 60 million years ago, either because they evolved via a different mechanism or deletions and substitutions have degraded the signal of TE involvement to the point at which they are no longer recognizable (Ellison, 2013).
The human orthologue of the tumor suppressor protein FBW7 is encoded by the Drosophila archipelago (ago) gene. Ago is an F-box protein that gives substrate specificity to its SCF ubiquitin ligase complex. It has a central role in multiple biological processes in a tissue-specific manner such as cell proliferation, cellular differentiation, hypoxia-induced gene expression. This study presents a previously unknown tissue-specific role of Ago in spermatid differentiation. A classical mutant of ago was identified that is semi-lethal and male-sterile. During the characterization of ago function in testis, ago was found to play role in spermatid development, following meiosis. Spermatogenesis defects was confirmed by silencing ago by RNAi in testes. The v mutants show multiple abnormalities in elongating and elongated spermatids, including aberration of the cyst morphology, malformed mitochondrial structures, and individualization defects. Additionally, the subcellular localization of Ago protein was determined with mCherry-Ago transgene in spermatids. These findings highlight the potential roles of Ago in different cellular processes of spermatogenesis, like spermatid individualization, and regulation of mitochondrial morphology (Vedelek, 2021).
Mechanisms of sex chromosome dosage compensation (SCDC) differ strikingly among animals. In Drosophila flies, chromosome-wide transcription is doubled from the single X chromosome in hemizygous (XY) males, whereas in Caenorhabditis nematodes, expression is halved for both X copies in homozygous (XX) females. Unlike other female-heterogametic (WZ female and ZZ male) animals, moths and butterflies exhibit sex chromosome dosage compensation patterns typically seen only in male-heterogametic species. The monarch butterfly carries a newly derived Z chromosome segment that arose from an autosomal fusion with the ancestral Z. Using a highly contiguous genome assembly, this study shows that gene expression is balanced between sexes along the entire Z chromosome but with distinct modes of compensation on the two segments. On the ancestral Z segment, depletion of H4K16ac corresponds to nearly halving of biallelic transcription in males, a pattern convergent to nematodes. Conversely, the newly derived Z segment shows a Drosophila-like mode of compensation, with enriched H4K16ac levels corresponding to doubled monoallelic transcription in females. This work reveals that, contrary to the expectation of co-opting regulatory mechanisms readily in place, the evolution of plural modes of dosage compensation is also possible along a single sex chromosome within a species (Gu, 2019).
Sex chromosomes often differ from autosomes with respect to their gene expression and regulation. In Drosophila melanogaster, X-linked genes are dosage compensated by having their expression up-regulated in the male soma, a process mediated by the X chromosome-specific binding of the dosage compensation complex (DCC). Previous studies of X-linked gene expression found a negative correlation between a gene's male-to-female expression ratio and its distance to the nearest DCC binding site in somatic tissues, including head and brain, which suggests that dosage compensation influences sex-biased gene expression. A limitation of the previous studies, however, was that they focused on endogenous X-linked genes and, thus, could not disentangle the effects of chromosomal position from those of gene-specific regulation. To overcome this limitation, this study examined the expression of an exogenous reporter gene inserted at many locations spanning the X chromosome. A negative correlation was observed between the male-to-female expression ratio of the reporter gene and its distance to the nearest DCC binding site in somatic tissues, but not in gonads. A reporter gene's location relative to a DCC binding site had greater influence on its expression than the local regulatory elements of neighboring endogenous genes, suggesting that intra-chromosomal variation in the strength of dosage compensation is a major determinant of sex-biased gene expression. Average levels of sex-biased expression did not differ between head and brain, but there was greater positional effect variation in the brain, which may explain the observed excess of endogenous sex-biased genes located on the X chromosome in this tissue (Belyi, 2020).
Given their copy number differences and unique modes of inheritance, the evolved gene content and expression of sex chromosomes is unusual. In many organisms the X and Y chromosomes are inactivated in spermatocytes, possibly as a defense mechanism against insertions into unpaired chromatin. In addition to current sex chromosomes, Drosophila has a small gene-poor X-chromosome relic (4(th)) that re-acquired autosomal status. This study used single cell RNA-Seq on fly larvae to demonstrate that the single X and pair of 4(th) chromosomes are specifically inactivated in primary spermatocytes, based on measuring all genes or a set of broadly expressed genes in testis that were identified. In contrast, genes on the single Y chromosome become maximally active in primary spermatocytes. Reduced X transcript levels are due to failed activation of RNA-Polymerase-II by phosphorylation of Serine 2 and 5 (Mahadevaraju, 2021).
Dosage compensation is a mechanism that equalizes sex chromosome gene expression between the sexes. In Drosophila, individuals with two X chromosomes (XX) become female, whereas males have one X chromosome (XY). In males, dosage compensation of the X chromosome in the soma is achieved by five proteins and two non-coding RNAs, which assemble into the male-specific lethal (MSL) complex to upregulate X-linked genes twofold. By contrast, it remains unclear whether dosage compensation occurs in the germline. To address this issue, transcriptome analysis was performed of male and female primordial germ cells (PGCs). The expression levels of X-linked genes were approximately twofold higher in female PGCs than in male PGCs. Acetylation of lysine residue 16 on histone H4 (H4K16ac), which is catalyzed by the MSL complex, was undetectable in these cells. In male PGCs, hyperactivation of X-linked genes and H4K16ac were induced by overexpression of the essential components of the MSL complex, which were expressed at very low levels in PGCs. Together, these findings indicate that failure of MSL complex formation results in the absence of X-chromosome dosage compensation in male PGCs (Ota, 2021).
Sex chromosomes induce potentially deleterious gene expression imbalances that are frequently corrected by dosage compensation (DC). Three distinct molecular strategies to achieve DC have been previously described in nematodes, fruit flies, and mammals. Is this a consequence of distinct genomes, functional or ecological constraints, or random initial commitment to an evolutionary trajectory? DC was studied in the malaria mosquito Anopheles gambiae. The Anopheles and Drosophila X chromosomes evolved independently but share a high degree of homology. Anopheles achieves DC by a mechanism distinct from the Drosophila MSL complex-histone H4 lysine 16 acetylation pathway. CRISPR knockout of Anopheles msl-2 leads to embryonic lethality in both sexes. Transcriptome analyses indicate that this phenotype is not a consequence of defective X chromosome DC. By immunofluorescence and ChIP, H4K16ac does not preferentially enrich on the male X. Instead, the mosquito MSL pathway regulates conserved developmental genes. It is concluded that a novel mechanism confers X chromosome up-regulation in Anopheles. These findings highlight the pluralism of gene-dosage buffering mechanisms even under similar genomic and functional constraints (Valsecchi, 2021).
Drosophila dosage compensation is an important model system for defining how active chromatin domains are formed. The male-specific lethal dosage compensation complex (MSLc) increases transcript levels of genes along the length of the single male X-chromosome to equalize with that expressed from the two female X-chromosomes. The strongest binding sites for MSLc cluster together in three-dimensional space largely independent of MSLc because clustering occurs in both sexes. CLAMP, a non-sex specific, ubiquitous zinc finger protein, binds synergistically with MSLc to enrich the occupancy of both factors on the male X-chromosome. This study demonstrates that CLAMP promotes the observed three-dimensional clustering of MSLc binding sites. Moreover, the X-enriched CLAMP protein more strongly promotes longer-range three-dimensional interactions on the X-chromosome than autosomes. Genome-wide, CLAMP promotes three-dimensional interactions between active chromatin regions together with other insulator proteins. This study has defined how long-range interactions which are modulated by a locally enriched ubiquitous transcription factor promote hyper-activation of the X-chromosome to mediate dosage compensation (Jordan, 2021).
Dosage compensation in Drosophila melanogaster involves a 2-fold transcriptional upregulation of the male X chromosome, which relies on the X-chromosome-binding males-specific lethal (MSL) complex (see msl-2). However, how such 2-fold precision is accomplished remains unclear. This study shows that a nuclear pore component, Mtor, is involved in setting the correct levels of transcription from the male X chromosome. Using larval tissues, this study demonstrated that the depletion of Mtor results in selective upregulation at MSL targets of the male X, beyond the required 2-fold. Mtor and MSL components interact genetically, and depletion of Mtor can rescue the male lethality phenotype of MSL components. Using RNA fluorescence in situ hybridization (FISH) analysis and nascent transcript sequencing, this study found that the effect of Mtor is not due to defects in mRNA export but occurs at the level of nascent transcription. These findings demonstrate a physiological role for Mtor in the process of dosage compensation, as a transcriptional attenuator of X chromosome gene expression (Aleman, 2021).
Dosage compensation
equalizes X-linked expression between XY males and XX females. In male
fruit flies, expression levels of the X-chromosome are increased
approximately two-fold to compensate for their single X chromosome. In testis,
dosage compensation is thought to cease during meiosis; however, the
timing and degree of the resulting transcriptional suppression is
difficult to separate from global meiotic downregulation of each
chromosome. To address this, testis single-cell RNA-sequencing
(scRNA-seq) data was analyzed from two Drosophila melanogaster strains.
Evidence was found that the X chromosome is equally transcriptionally
active as autosomes in somatic and pre-meiotic cells, and less
transcriptionally active than autosomes in meiotic and post-meiotic
cells. In cells experiencing dosage compensation, close proximity to MSL
(male-specific lethal) chromatin entry sites (CES) correlates with
increased X chromosome transcription. Low or undetectable levels of
germline expression of most msl genes, mle, roX1 and roX2 were found via
scRNA-seq and RNA-FISH, and no evidence was found of germline nuclear
roX1/2 localization. These results suggest that, although dosage
compensation occurs in somatic and pre-meiotic germ cells in Drosophila
testis, there might be non-canonical factors involved in the dosage
compensation mechanism. The single-cell expression patterns and
enrichment statistics of detected genes can be explored interactively in
a database (Witt, 2021).
The dosage compensation complex (DCC)
of Drosophila identifies its X-chromosomal binding sites with exquisite
selectivity. The principles that assure this vital targeting are known
from the D. melanogaster model: DCC-intrinsic specificity of DNA
binding, cooperativity with the CLAMP protein, and noncoding roX2
RNA transcribed from the X chromosome. This study found that in D.
virilis, a species separated from melanogaster by 40 million years of
evolution, all principles are active but contribute differently to X
specificity. In melanogaster, the DCC subunit MSL2 evolved intrinsic
DNA-binding selectivity for rare PionX sites, which mark the X
chromosome. In virilis, PionX motifs are abundant and not X-enriched.
Accordingly, MSL2 lacks specific recognition. Here, roX2 RNA plays a
more instructive role, counteracting a nonproductive interaction of
CLAMP and modulating DCC binding selectivity. Remarkably, roX2 triggers a
stable chromatin binding mode characteristic of DCC. Evidently, X-specific regulation is achieved by divergent evolution of protein, DNA, and RNA components (Villa, 2021).
Dosage compensation (DC), a process countering chromosomal imbalance in individuals with heteromorphic sex chromosomes, has been molecularly characterized only in mammals, Caenorhabditis elegans, and fruit flies. In Drosophila melanogaster males, it is achieved by an approximately 2-fold hypertranscription of the monosomic X chromosome mediated by the MSL complex. The complex is not assembled on female X chromosomes because production of its key protein MSL-2 is prevented due to intron retention and inhibition of translation by Sex-lethal, a female-specific protein operating at the top of the sex determination pathway. It remains unclear how DC is mechanistically regulated in other insects. In the malaria mosquito Anopheles gambiae, an approximately 2-fold hypertranscription of the male X also occurs by a yet-unknown molecular mechanism distinct from that in D. melanogaster. This study shows that a male-specifically spliced gene called 007, which arose by a tandem duplication in the Anopheles ancestral lineage, is involved in the control of DC in males. Homozygous 007 knockouts lead to a global downregulation of the male X, phenotypically manifested by a slower development compared to wild-type mosquitoes or mutant females-however, without loss of viability or fertility. In females, a 007 intron retention promoted by the sex determination protein Femaleless, known to prevent hypertranscription from both X chromosomes, introduces a premature termination codon apparently rendering the female transcripts non-productive. In addition to providing a unique perspective on DC evolution, the 007, with its conserved properties, may represent an important addition to a genetic toolbox for malaria vector control (Krzywinska, 2023).
Sex chromosomes originated from autosomes but have evolved a highly specialized chromatin structure. Drosophila Y chromosomes are composed entirely of silent heterochromatin, while male X chromosomes have highly accessible chromatin and are hypertranscribed as a result of dosage compensation. This study dissected the molecular mechanisms and functional pressures driving heterochromatin formation and dosage compensation of the recently formed neo-sex chromosomes of Drosophila miranda. The onset of heterochromatin formation on the neo-Y is triggered by an accumulation of repetitive DNA. The neo-X has evolved partial dosage compensation and it was found that diverse mutational paths have been utilized to establish several dozen novel binding consensus motifs for the dosage compensation complex on the neo-X, including simple point mutations at pre-binding sites, insertion and deletion mutations, microsatellite expansions, or tandem amplification of weak binding sites. Spreading of these silencing or activating chromatin modifications to adjacent regions results in massive mis-expression of neo-sex linked genes, and little correspondence between functionality of genes and their silencing on the neo-Y or dosage compensation on the neo-X. Intriguingly, the genomic regions being targeted by the dosage compensation complex on the neo-X and those becoming heterochromatic on the neo-Y show little overlap, possibly reflecting different propensities along the ancestral chromosome that formed the sex chromosome to adopt active or repressive chromatin configurations. These findings have broad implications for current models of sex chromosome evolution, and demonstrate how mechanistic constraints can limit evolutionary adaptations. The study also highlights how evolution can follow predictable genetic trajectories, by repeatedly acquiring the same 21-bp consensus motif for recruitment of the dosage compensation complex, yet utilizing a diverse array of random mutational changes to attain the same phenotypic outcome (Zhou, 2013).
D. miranda has a unique karyotype, harboring three sex chromosomes of different ages: XL is the ancestral X chromosome in the genus Drosophila and >60 MY old, XR became X-linked about 15 MY ago, is entirely dosage compensated and its former homolog is completely degenerated (i.e., all genes on XR are hemizygous in males). This implies that a former autosome can become completely transformed into a heteromorphic sex chromosome within only 15 MY in Drosophila. The much younger neo-sex chromosomes are at an earlier stage of this evolutionary transition, and the neo-Y is only partially degenerated and the neo-X has evolved incomplete dosage compensation. This provides a unique opportunity to study the evolutionary processes driving the differentiation of sex chromosomes, and this study investigated how changes to the DNA sequence result in novel epigenetic modifications of the diverging neo-sex chromosomes that affect levels of transcription of neo-sex linked genes. Recruitment of the dosage compensation complex to the neo-X requires the acquisition of a 21-bp consensus motif, and this study uncovered diverse mutational paths that have led to the evolution of novel CES on the neo-X. This highlights how evolution can follow predictable genetic trajectories by repeatedly acquiring the same 21-bp consensus motif for recruitment of the dosage compensation complex, yet utilizing a diverse array of random mutational changes to attain the same phenotypic outcome. It was further shown that heterochromatin formation is triggered by an accumulation of repetitive DNA on the neo-Y, and silences adjacent genes (Zhou, 2013).
Highly differentiated sex chromosomes create a lethal imbalance in gene expression in one sex. To accommodate hemizygosity of the X chromosome in male fruit flies, expression of X-linked genes increases twofold. This is achieved by the male- specific lethal (MSL) complex, which modifies chromatin to increase expression. Mutations that disrupt the X localization of this complex decrease the expression of X-linked genes and reduce male survival. The mechanism that restricts the MSL complex to X chromatin is not understood. The siRNA pathway has been shown to contribute to localization of the MSL complex, raising questions about the source of the siRNAs involved. The X-linked 1.688 g/cm3 satellite related repeats (1.688X repeats; 359-bp repeat unit) are restricted to the X chromosome and produce small RNA, making them an attractive candidate. RNA from these repeats was tested for a role in dosage compensation, and ectopic expression of single-stranded RNAs from 1.688X repeats was found to enhance the male lethality of mutants with defective X recognition. In contrast, expression of double-stranded hairpin RNA from a 1.688X repeat generated abundant siRNA and dramatically increased male survival. Consistent with improved survival, X localization of the MSL complex was largely restored in these males. The striking distribution of 1.688X repeats, which are nearly exclusive to the X chromosome, suggests that these are cis-acting elements contributing to identification of X chromatin (Menon, 2014: PubMed).
Dosage compensation mechanisms provide a paradigm to study the contribution
of chromosomal conformation toward targeting and spreading of epigenetic
regulators over a specific chromosome. By using Hi-C and 4C analyses,
this study shows that high-affinity sites (HAS), landing platforms of
the male-specific lethal (MSL)
complex, are enriched around topologically associating domain (TAD)
boundaries on the X chromosome and harbor more long-range contacts in a
sex-independent manner. Ectopically expressed roX1 and roX2 RNAs target HAS on the X
chromosome in trans and, via spatial proximity, induce spreading of the
MSL complex in cis, leading to increased expression of neighboring
autosomal genes. It was shown that the MSL complex regulates nucleosome
positioning at HAS, therefore acting locally rather than influencing the
overall chromosomal architecture. The study proposes that the
sex-independent, three-dimensional conformation of the X chromosome
poises it for exploitation by the MSL complex, thereby facilitating
spreading in males (Ramírez, 2015).
This study provides a first step toward understanding the role of chromosome conformation in dosage compensation in D. melanogaster. HAS, the landing regions of the MSL complex on the X chromosome, frequently reside in proximity to TAD boundaries. HAS are enriched in Hi-C contacts to each other and to other X chromosomal regions and that this organization remains comparable between male and female cells (Ramírez, 2015).
This analysis revealed that HAS are characterized by a combination of DNA sequence (MREs), chromatin state (active), and gene architecture, which drives the specificity of the MSL complex toward the X chromosome. The data suggest that when the MSL complex binds to HAS, it then spreads (either via an active mechanism or via diffusion) to spatially close regions to place the histone H4 lysine 16 acetylation (H4K16ac) mark on active genes. A 'conformation-based affinity' model is proposed based on the strategic location of HAS at highly interconnected regions of the D. melanogaster X chromosome that efficiently distribute the MSL complex over the X chromosome by attracting the MSL complex to cis-interacting HAS on the X chromosome. This system ensures that only this chromosome is specifically and globally targeted. By spreading from those HAS over short (3D) distances, all active genes on the X chromosome are then reached and acetylated without influencing the autosomes. It is suggested that this system is resilient to major perturbations, exemplified by the large autosomal insertion from chromosome 3L and the ectopic expression of the roX genes that produce viable cells and flies, respectively (Ramírez, 2015).
MNase-seq analysis shows a direct effect of the MSL complex on nucleosome organization specifically on HAS and not on the TSS, despite prominent binding of MSL1/2 to promoter regions. The MSL complex may act similar to a pioneer DNA binding protein to establish nucleosome patterns at HAS and may act on neighboring active regions rather than modifying TAD boundaries. This system may be unique to flies because the Drosophila dosage compensation evolved a fine-tuning transcription activation mechanism rather than a complete shutdown of gene transcription as seen in mammalian X chromosome inactivation. It would be very interesting to see how nucleosome positioning is affected upon Xist binding in mammals (Ramírez, 2015).
Although many factors, including the CCCTC-binding factor (CTCF) as well as tRNA and housekeeping genes, have been shown to be enriched at boundaries, by dissecting the targeting and spreading activity of the MSL complex for the X chromosome, this study offers a plausible explanation behind the advantages of HAS localization. HAS are enriched at the X chromosomal boundaries and not at autosomal boundaries, where all other boundary factors will bind indiscriminately. Furthermore, it was found that the few HAS that are not near a boundary also occupy locations of an elevated number of long-range contacts, indicating that HAS form interaction hubs for the spreading of the MSL complex (Ramírez, 2015).
Hi-C as well as in vivo immunofluorescence show that active roX genes have more contacts and are closer to each other than inactive regions. These observations are in line with previous reports showing that active chromatin compartments interact more often with each other and that active chromatin localizes to the interface of the chromosomal territory. The results imply that different transcriptional programs in each cell line or tissue are likely to be associated with a particular arrangement of long-range contacts, suggesting that the dosage compensation must be flexible to act over such diverse conformations without disturbing them. This idea is consistent with the observation that the chromosome conformation remains unchanged after knockdown of the MSL complex, and stays in contrast to mammalian X inactivation, which involves chromatin condensation, gene inactivation, and alterations in chromosome conformation (Ramírez, 2015).
Dosage compensation mechanisms in flies and mammals lead to opposite outcomes; namely, gene activation versus gene repression. However, both systems use lncRNAs transcribed from the dosage-compensated X chromosome. roX1 and roX2 RNA are expressed from the male hyperactivated X chromosome in D. melanogaster, whereas Xist is expressed from the inactivated X chromosome in mammalian females. Recent work has shown that Xist spreads to distal sites on the X chromosome. Interestingly, this spreading is dependent on the spatial proximity of sites distal to the Xist gene. This is further exemplified by ectopic expression of Xist from chromosome 21, where Xist spread only in cis on this chromosome. In this study, ectopic insertion of roX transgenes on autosomes demonstrated that the roX/MSL complex can reach the X chromosome and rescue male lethality. Therefore, acting in trans is a special feature of roX RNAs (in conjunction with the MSL complex) not observed for Xist, indicating that the two systems utilize the respective lncRNAs differently. In both systems, however, the lncRNAs need to be functional because the stem loop structures of the roX RNAs are required for dosage compensation in D. melanogaster, whereas Xist needs the "A repeat domain" to induce mammalian X chromosome inactivation. The distinct mechanisms utilized by the Xist and roX RNAs exemplify the great versatility by which lncRNAs can be involved in the global regulation of single chromosomes and might reflect important differences between the two systems. In mammals, only one of the two X chromosomes needs to be inactivated. Therefore, a trans action of Xist RNA on the sister X chromosome would be detrimental to the organism. In contrast, the dosage-compensated X chromosome is present singularly in males in Drosophila. However, because the roX RNAs can act in trans, it may be disadvantageous to target the activating MSL complex to active genes on autosomes, hence the need for specific target regions (the HAS) unique to the X chromosome (Ramírez, 2015).
To fully understand the occurrence of HAS at sites with extensive long-range interactions on the X chromosomes, it could be helpful to consider evolutionary models proposing that X chromosomes tend to evolve faster than autosomes (faster X effect). Under the faster X effect, traits only beneficial for males can introduce significant changes specific to the X chromosome on a short evolutionary timescale. Based on these and other observations suggesting that the X chromosome in flies is different from autosomes, it is assumed that selective pressures on males favored the occurrence of HAS at regions of increased interactions, like TAD boundaries. Future analyses of different Drosophila species will open exciting opportunities to study the evolutionary changes of HAS in the context of X chromosomal architecture. Moreover, conformation-based affinity could be a generic mechanism for other regulatory elements to exert their functions. It remains to be seen in which contexts the in cis versus in trans action of different lncRNAs is essential for their function and how chromosome conformation, long-range contacts, HAS, and regulation of transcription have co-evolved for dosage compensation (Ramírez, 2015).
The ribonucleoprotein Male Specific Lethal (MSL) complex is required for X chromosome dosage compensation in Drosophila males. Beginning at 3 h of development the MSL complex binds transcribed X-linked genes and modifies chromatin. A subset of MSL complex proteins, including MSL1 and MSL3, is also necessary for full expression of autosomal heterochromatic genes in males, but not females. Loss of the non-coding roX RNAs, essential components of the MSL complex, lowers the expression of heterochromatic genes and suppresses position effect variegation (PEV) only in males, revealing a sex-limited disruption of heterochromatin. MLE, but not Jil-1 kinase, was found to contribute to heterochromatic gene expression. To determine if identical regions of roX RNA are required for dosage compensation and heterochromatic silencing, a panel of roX1 transgenes and deletions was tested; the X chromosome and heterochromatin functions were found to be separable by some mutations. Widespread autosomal binding of MSL3 occurs before and after localization of the MSL complex to the X chromosome at 3 h AEL. Autosomal MSL3 binding was dependent on MSL1, supporting the idea that a subset of MSL proteins associates with chromatin throughout the genome during early development. It is postulated that this binding may contribute to the sex-specific differences in heterochromatin that have been noted (Koya, 2015).
A central question raised by this study is how factors known for their role in X chromosome dosage compensation also modulate autosomal heterochromatin. Although the MSL proteins were first identified by their role in X chromosome compensation, homologues of these proteins participate in chromatin organization, DNA repair, gene expression, cell metabolism and neural function throughout the eukaryotes. Furthermore, flies contain a distinct complex, the Non-Sex specific Lethal (NSL) complex, containing MOF and the MSL orthologs NSL1, NSL2 and NSL3. The essential NSL complex is broadly associated with promoters throughout the fly genome, where it acetylates multiple H4 residues. In light of the discovery that the MSL proteins represent an ancient lineage of chromatin regulators, it is unsurprising that members of this complex fulfill additional functions (Koya, 2015).
An alternative hypothesis for the dosage compensation of male X-linked genes proposes that the MSL proteins are general transcription regulators, and recruitment of these factors to the male X chromosome reduces autosomal gene expression, thus equalizing the X:A expression ratio. Arguing against this idea are ChIP studies finding that the MSL complex, and engaged RNA polymerase II, are increased within the bodies of compensated X-linked genes. In agreement with this, a study that normalized expression to genomic DNA concluded that compensation increases the expression of male X-linked genes. The current study now reveals that autosomal heterochromatic genes are indeed dependent on a subset of MSL proteins for full expression. However, native heterochromatic genes make up only 4% of autosomal genes, and their misregulation is not expected to compromise genome-wide expression studies normalized to autosomal expression (Koya, 2015).
Expression of heterochromatic genes is thought to involve mechanisms to overcome the repressive chromatin environment. It is possible that a complex composed of roX RNA and a subset of MSL proteins participates in this process. This would explain why heterochromatic genes are particularly sensitive to the loss of these factors. Alternatively, it is possible that roX and MSL proteins participate in heterochromatin assembly. This would explain the simultaneous disruption of heterochromatic gene expression and suppression of PEV at transgene insertions (Koya, 2015).
Heterochromatin assembly is first detected at 3-4 h AEL, a time when MSL3 is bound throughout the genome. Intriguingly, studies from yeast identify a role for H3K4 and H4K16 acetylation in formation of heterochromatin. Active deacetylation of H4K16ac is necessary for spreading of chromatin-based silencing in yeast, demonstrating the need for a sequential and ordered series of histone modifications (Koya, 2015).
As MOF is responsible for the majority of H4K16ac in the fly, a MOF-containing complex could fulfill a similar role during heterochromatin formation. While this study found a significant effect of MOF in expression only on the X and 4th chromosomes, it is possible that examination of a larger number of genes would reveal a more widespread autosomal effect (Koya, 2015).
In roX1 roX2 males the 4th chromosome displays stronger suppression of PEV and more profound gene misregulation than do other heterochromatic regions. This is consistent with the observation that heterochromatin on the 4th chromosome is genetically and biochemically different from that on other chromosomes. Loss of roX RNA leads to misregulation of genes in distinct genomic regions, the dosage compensated X chromosome and autosomal heterochromatin. This study found that the regulation of these two groups is, to some extent, genetically separable. MSL2, which binds roX1 RNA and is an essential member of the dosage compensation complex, is not required for full expression of heterochromatic genes in males. Ectopic expression of MSL2 in females induces formation of MSL complexes that localize to both X chromosomes, inducing inappropriate dosage compensation. As would be expected from the lack of a role for MSL2 in autosomal heterochromatin in males, ectopic expression of this protein in females has no effect on PEV (Koya, 2015).
Elegant, high-resolution studies reveal that MLE and MSL2 bind essentially indistinguishable regions of roX1. Three prominent regions of MLE/MSL2 binding have been identified, one overlapping the 3' stem loop. This stem loop incorporates a short 'roX box' consensus sequence that is present in D. melanogaster roX1 and roX2, and conserved in roX RNAs in related species (Koya, 2015).
An experimentally supported explanation for the concurrence of MLE and MSL2 binding at the 3' stem loop is that MLE, an ATP-dependent RNA/DNA helicase, remodels this structure to permit MSL2 binding. The finding that disruption of this stem blocks dosage compensation but does not influence heterochromatic integrity is consistent with participation of roX1 in two processes that differ in MSL2 involvement. However, a region surrounding the stem loop is required for the heterochromatic function of roX1, as roX1Δ10, removing the stem loop and upstream regions, is deficient in both dosage compensation and heterochromatic silencing. Further differentiating these processes is the finding that low levels of roX RNA from a repressed transgene fully rescue heterochromatic silencing, but not dosage compensation. An intriguing question raised by this study is why the sexes display differences in autosomal heterochromatin (Koya, 2015).
The chromatin content of males and females are substantially different as XY males have a single X and a large, heterochromatic Y chromosome. It is speculated that this has driven changes in how heterochromatin is established or maintained in one sex. A search for the genetic regulators of the sex difference in autosomal heterochromatin eliminated the Y chromosome and the conventional sex determination pathway, suggesting that the number of X chromosomes determines the sensitivity of autosomal heterochromatin to loss of roX activity. Interestingly, the amount of pericentromeric X heterochromatin, rather than the euchromatic 'numerator' elements, appears to be the critical factor. The recognition that heterochromatin displays differences in the sexes, and that a specific set of proteins are required for normal function of autosomal heterochromatin in males suggests a useful paradigm for the evolution of chromatin in response to genomic content (Koya, 2015).
Numerous arthropods harbor maternally transmitted bacteria that induce the preferential death of males. This sex-specific lethality benefits the bacteria because males are "dead ends" regarding bacterial transmission, and their absence may result in additional resources for their viable female siblings who can thereby more successfully transmit the bacteria. Although these symbionts disrupt a range of developmental processes, the underlying cellular mechanisms are largely unknown. It has previously been shown that mutations in genes of the dosage compensation pathway of Drosophila melanogaster suppress male killing caused by the bacterium Spiroplasma. This suggests that dosage compensation is a target of Spiroplasma. However, it remains unclear how this pathway is affected, and whether the underlying interactions require the male-specific cellular environment. This study investigated the cellular basis of male embryonic lethality in D. melanogaster induced by Spiroplasma. It was found that the dosage compensation complex (DCC), which acetylates X chromatin in males, becomes mis-localized to ectopic regions of the nucleus immediately prior to the killing phase. This effect is accompanied by inappropriate histone acetylation and genome-wide mis-regulation of gene expression. Artificially induced formation of the DCC in infected females, through transgenic expression of the DCC-specific gene msl-2, results in mis-localization of this complex to non-X regions and early Spiroplasma-induced death, mirroring the killing effects in males. These findings strongly suggest that Spiroplasma initiates male killing by targeting the dosage compensation machinery directly and independently of other cellular features characteristic of the male sex (Cheng, 2016)
In the male germline of Drosophila melanogaster, a novel but poorly understood form of sex chromosome-specific transcriptional regulation occurs that is distinct from canonical sex chromosome dosage compensation or meiotic inactivation. Previous work shows that expression of reporter genes driven by testis-specific promoters is considerably lower-approximately 3-fold or more-for transgenes inserted into X chromosome versus autosome locations. This study characterized transcriptional suppression of X-linked genes in the male germline and its evolutionary consequences. Using transgenes and transpositions, most endogenous X-linked genes, not just testis-specific ones, were shown to be transcriptionally suppressed several-fold specifically in the Drosophila male germline. In wild-type testes, this sex chromosome-wide transcriptional suppression is generally undetectable, being effectively compensated by the gene-by-gene evolutionary recruitment of strong promoters on the X chromosome. A promoter element sequence motif was was identified and experimentally validated that is enriched upstream of the transcription start sites of hundreds of testis-expressed genes; evolutionarily conserved across species; associated with strong gene expression levels in testes; and overrepresented on the X chromosome. These findings show that the expression of X-linked genes in the Drosophila testes reflects a balance between chromosome-wide epigenetic transcriptional suppression and long-term compensatory adaptation by sex-linked genes (Landeen, 2016).
Heterogametic species require chromosome-wide gene regulation to compensate for differences in sex chromosome gene dosage. In Drosophila melanogaster, transcriptional output from the single male X-chromosome is equalized to that of XX females by recruitment of the male-specific lethal (MSL) complex, which increases transcript levels of active genes 2-fold. The MSL complex contains several protein components and two non-coding RNA on the X (roX) RNAs that are transcriptionally activated by the MSL complex. Targeting of the MSL complex to the X-chromosome has been shown to be dependent on the chromatin-linked adapter for MSL proteins (CLAMP) zinc finger protein. To better understand CLAMP function, the CRISPR/Cas9 genome editing system was used to generate a frameshift mutation in the clamp gene that eliminates expression of the CLAMP protein. clamp null females were found to die at the third instar larval stage, while almost all clamp null males die at earlier developmental stages. Moreover, it was found that in clamp null females roX gene expression is activated, whereas in clamp null males roX gene expression is reduced. Therefore, CLAMP regulates roX abundance in a sex-specific manner. These results provide new insights into sex-specific gene regulation by an essential transcription factor (Urban, 2017).
Many species employ a sex determination system that generates an inherent imbalance in sex chromosome copy number, such as the XX/XY system in most mammals and some insects. In this system, one sex has twice the number of X-chromosome-encoded genes compared to the other. Therefore, a mechanism of dosage compensation is required to equalize levels of X-linked transcripts, both between the sexes and between the X-chromosome and autosomes. Dosage compensation is an essential mechanism that corrects for this imbalance by coordinately regulating the gene expression of most X-linked genes (Urban, 2017).
In Drosophila melanogaster, transcription from the single male X-chromosome is increased 2-fold by recruitment of the male-specific lethal (MSL) complex. The MSL complex is composed of two structural proteins, MSL1 and MSL2, three accessory proteins, MSL3, males absent on the first (MOF), and maleless (MLE), and two functionally redundant non-coding RNAs, RNA on the X (roX1) and roX2. Previous work has shown that recruitment of the MSL complex to the X-chromosome requires the zinc finger protein chromatin-linked adapter for MSL proteins (CLAMP) (Soruco, 2013; Urban, 2017 and references therein).
In addition to its role in male MSL complex recruitment, it was suggested that CLAMP has an additional non-sex-specific essential function because targeting of the clamp transcript by RNA interference results in a pupal lethal phenotype in both males and females (Soruco, 2013). Further understanding of CLAMP function in the context of the whole organism required a null mutant. However, due to the pericentric location of the clamp gene, no deficiencies or null mutations were available. Using the CRISPR/Cas9 system, a frameshift mutation was introduced in the clamp gene, leading to an early termination codon before the major zinc finger binding domain. This frameshift mutation generated the clamp2 allele, which eliminates detectable CLAMP protein production and is therefore a protein null allele. The majority of clamp2 mutant males die prior to the third instar stage. On the other hand, females die at the third instar stage, suggesting sex-specific functions for CLAMP. Furthermore, CLAMP regulates the roX genes in a sex-specific manner, activating their accumulation in
males and repressing their accumulation in females. Overall, we present a new tool for studying dosage compensation and suggest that CLAMP functions to assure that roX RNA accumulation is sex specific (Urban, 2017).
Previous work demonstrated that CLAMP has an essential role in MSL complex recruitment to the male X-chromosome (Soruco, 2013). However, it was not possible to perform in vivo studies to further investigate CLAMP function because there was no available null mutant line. The current work present a CLAMP protein null mutant and determine that this protein is essential in both sexes. This allele will provide a key tool for future in vivo studies on the role of CLAMP in dosage compensation, as well as identification of the essential function of CLAMP in both sexes (Urban, 2017).
The initial characterization of the clamp2 protein null allele revealed sexually dimorphic roles for CLAMP in regulation of the roX genes. CLAMP was seen to promotes roX2 transcription in males but represses transcription of both roX genes in females. It is likely that recruitment of the MSL complex to the roX2 locus by CLAMP promotes roX2 expression in males. In females, where the MSL complex is not present, CLAMP may function to repress these loci as an additional mechanism to ensure that dosage compensation is male-specific. Additionally, it was determined that most clamp2 homozygous males die earlier in development than clamp2 homozygous females. Earlier lethality in males is likely due to a misregulation of the dosage compensation process as a result of the loss of CLAMP-mediated MSL complex recruitment. However, CLAMP is enriched at the 5' regulatory regions of thousands of genes across the genome. Therefore, it is likely that other non-sex-specific regulatory pathways are disrupted resulting in female lethality (Urban, 2017).
Furthermore, CLAMP is an essential protein because our CRISPR/Cas9-generated protein null clamp allele is
homozygous lethal in both males and females. These results indicate that CLAMP has a previously unstudied non-sex-specific role that is essential to the viability of both males and females. An interesting observation that arose from this characterization is that polytene chromosome organization is disrupted in clamp2 mutant females, suggesting that CLAMP may play a role in regulation of genome-wide chromatin organization of interphase chromosomes. A function in regulating chromatin organization provides one possible explanation for how CLAMP performs sexually dimorphic functions. For example, CLAMP may repress roX expression in females by promoting the recruitment of a repressive chromatin-modifying factor in the absence of the MSL complex. In contrast, CLAMP may activate roX2 in males by creating a chromatin environment permissive for MSL complex recruitment in males. Although roX1 and roX2 are functionally redundant, the results suggest that CLAMP specifically activates roX2 but not roX1 in males. Interestingly, Villa (2016) recently reported that roX2, but not roX1, is likely to be an early site of MSL complex recruitment (Villa, 2016), suggesting that CLAMP may function early in the process of dosage compensation (Urban, 2017).
Overall, the newly generated clamp2 protein null allele provides an important tool to study how the essential CLAMP protein regulates its many target genes in vivo. The generation of the clamp2 allele will facilitate future studies that will reveal a mechanistic understanding of how a single transcription factor can promote different sex-specific functions within an organism (Urban, 2017).
Transcriptional regulators select their targets from a large pool of similar genomic sites. The binding of the Drosophila dosage compensation complex (DCC) exclusively to the male X chromosome provides insight into binding site selectivity rules. Previous studies showed that the male-specific organizer of the complex, MSL2, and ubiquitous DNA-binding protein CLAMP directly interact and play an important role in the specificity of X chromosome binding. The highly specific interaction between the intrinsically disordered region of MSL2 and the N-terminal zinc-finger C2H2-type (C2H2) domain of CLAMP was examined in this study. The NMR structure was obtainted of the CLAMP N-terminal C2H2 zinc finger, which has a classic C2H2 zinc-finger fold with a rather unusual distribution of residues typically used in DNA recognition. Substitutions of residues in this C2H2 domain had the same effect on the viability of males and females, suggesting that it plays a general role in CLAMP activity. The N-terminal C2H2 domain of CLAMP is highly conserved in insects. However, the MSL2 region involved in the interaction is conserved only within the Drosophila genus, suggesting that this interaction emerged during the evolution of a mechanism for the specific recruitment of the DCC on the male X chromosome in Drosophilidae (Tikhonova, 2022).
Dosage compensation is an essential process that equalizes transcript levels of X-linked genes between sexes by forming a domain of coordinated gene expression. Throughout the evolution of Diptera, many different X-chromosomes acquired the ability to be dosage compensated. Once each newly evolved X-chromosome is targeted for dosage compensation in XY males, its active genes are upregulated two-fold to equalize gene expression with XX females. In Drosophila melanogaster, the Chromatin-linked adaptor for MSL proteins (CLAMP) zinc finger protein links the dosage compensation complex to the X-chromosome. However, the mechanism for X-chromosome identification has remained unknown. This study combine biochemical, genomic and evolutionary approaches to reveal that expansion of GA-dinucleotide repeats likely accumulated on the X-chromosome over evolutionary time to increase the density of CLAMP binding sites, thereby driving the evolution of dosage compensation. Overall, this study presents new insight into how subtle changes in genomic architecture, such as expansions of a simple sequence repeat, promote the evolution of coordinated gene expression (Kuzu, 2016).
Upon the evolution of heterogametic species, the process of dosage compensation became essential to ensure the appropriate balance of gene expression between males and females and the X and autosomes. Distinguishing the X-chromosome from autosomes is the key step in this process because MSL complex must be targeted to the correct chromosome to ensure the fidelity of dosage compensation. This study has demonstrate that in several species this process likely involved enriching the evolving X-chromosomes for long GA-repeat binding sites that can be recognized by the highly conserved CLAMP protein that recruits MSL complex (Kuzu, 2016).
CLAMP binding sites are not X-specific as the CLAMP protein binds to similar GA-rich sequences all over the genome. It is proposed that a higher density of sites within CES that contain longer GA-repeats evolved to optimize CLAMP binding on X to better target MSL complex for dosage compensation. Then, it is likely that the increased density of CLAMP at CES functions together with other cofactors with known roles in MSL complex recruitment such as H3K36me3 and roX RNAs. Once this initial process of X-chromosome identification occurs, synergistic interactions between maternally loaded CLAMP and the MSL complex [20] increase the X-enrichment of both factors (Kuzu, 2016).
Interestingly, the CLAMP motif is much longer than most transcription factor binding sites. It is possible that the length of the CLAMP binding site ensures specificity by reducing the promiscuity of its binding and allowing it to compete with other similar proteins. In addition, recent work on transcriptional regulators in budding yeast has implicated the sequence context of transcription factor binding sites outside of the core binding site as critical for the recognition process. Therefore, current approaches to identifying transcription factor binding site motifs have likely underestimated their length due to the approaches used that often allow detection of only short motifs. In the future, it will be important to determine transcription factor recognition motifs using approaches like gcPBM that uses in vivo sequences to identify direct binding site motifs (Kuzu, 2016).
There are several mechanisms by which the GA-repeat number could have been increased including expansions due to slippage of DNA polymerase. Helitron transposons containing GA-rich sequences have also been implicated in the X-enrichment of these sequences in D. miranda. It is possible that expansions of GA dinucleotides occurred within these transposons after they landed on the X-chromosome. These GA-repeat expansions could have been further propagated by gene conversion events that also occurred during the evolution of dosage compensation. Finally, long repeat sequences such as the 1.688 elements that produce siRNAs function during dosage compensation via an unknown mechanism (Menon, 2014). Therefore, it is possible that GA-repeat elements have been expanded over evolutionary time because of a general role in promoting dosage compensation. To support this hypothesis, a recent report identified GA-rich binding motifs almost identical to those that we characterized as CLAMP binding sites within the strongest MSL complex binding sites in three additional Drosophila species (Kuzu, 2016).
Motifs that contain GA-repeats have been implicated in diverse processes that all involve generating open chromatin regions. GA-repeat containing motifs are highly enriched at sites that promote pausing of RNA Polymerase II and at developmentally regulated DNase I hypersensitivity sites. Furthermore, a GA-repeat motif is one of the two motifs that are enriched at genes that are activated first during the maternal to zygotic transition. The well-studied GAGA factor (GAF) protein also recognizes similar sequences to the CLAMP protein and has been implicated in pausing of RNA Polymerase II and opening of chromatin. Overall, it is likely that the dosage compensation machinery has evolved to take advantage of targeting GA-repeats that mark open chromatin regions to ensure that it only identifies active genes for further transcriptional upregulation by the MSL complex (Kuzu, 2016).
It is possible that GA-rich sequences have roles in dosage compensation outside of Diptera. For example, it has been proposed that upregulation of the single active X occurs in mammals and this process is mediated by targeting the conserved MOF histone acetyltransferase component of MSL complex. Moreover, GA-repeats were found to be significantly enriched within regions of the X-chromosome that escape X-inactivation (X escape regions). There are no strong homologues of CLAMP in mammals but there are several possible functional orthologs such as the ETS family transcription factor GABP1 (GA binding protein-1). Furthermore, in C. elegans, there is an early upregulation of both X-chromosomes that is also mediated by the MOF histone acetyltransferase. One of the zinc finger proteins that targets the C. elegans dosage compensation machinery is SCC-2 (sister chromatid cohesion-2) which recognizes a GA-repeat sequence very similar to the CLAMP binding motif. Therefore, it is possible that GA-repeats are involved in dosage compensation beyond Diptera and this will be an exciting area for future investigation (Kuzu, 2016).
Deletions, commonly referred to as deficiencies by Drosophila
geneticists, are valuable tools for mapping genes and for genetic pathway
discovery via dose-dependent suppressor and enhancer screens. More
recently, it has become clear that deviations from normal gene dosage are
associated with multiple disorders in a range of species including humans.
While some of the transcriptional effects brought about by gene dosage
changes and the chromosome rearrangement breakpoints associated with them
are beginning to be understood, much of this work relies on isolated
examples. This study systematically examined deficiencies of the left arm
of chromosome 2 and characterized gene-by-gene dosage responses that vary
from collapsed expression through modest partial dosage compensation to
full or even over compensation. Negligible long-range effects of creating
novel chromosome domains at deletion breakpoints were found, suggesting
that cases of gene regulation due to altered nuclear architecture are
rare. These rare cases include trans de-repression when deficiencies
delete chromatin characterized as repressive in other studies. Generally,
effects of breakpoints on expression are promoter proximal (~100bp) or in
the gene body. Effects of deficiencies genome-wide are in genes with
regulatory relationships to genes within the deleted segments,
highlighting the subtle expression network defects in these sensitized
genetic backgrounds (Lee, 2016). The rules defining which small fraction of related DNA sequences can be selectively bound by a transcription factor are poorly understood. One of the most challenging tasks in DNA recognition is posed by dosage compensation systems that require the distinction between sex chromosomes and autosomes. In Drosophila melanogaster, the male-specific lethal dosage compensation complex (MSL-DCC) doubles the level of transcription from the single male X chromosome, but the nature of this selectivity is not known. Previous efforts to identify X-chromosome-specific target sequences were unsuccessful as the identified MSL recognition elements lacked discriminative power. Therefore, additional determinants such as co-factors, chromatin features, RNA and chromosome conformation have been proposed to refine targeting further. Using an in vitro genome-wide DNA binding assay this study shows that recognition of the X chromosome is an intrinsic feature of the MSL-DCC. MSL2, the male-specific organizer of the complex, uses two distinct DNA interaction surfaces-the CXC and proline/basic-residue-rich domains-to identify complex DNA elements on the X chromosome. Specificity is provided by the CXC domain, which binds a novel motif defined by DNA sequence and shape. This motif characterizes a subclass of MSL2-binding sites, which has been named PionX (pioneering sites on the X) as they appeared early during the recent evolution of an X chromosome in D. miranda and are the first chromosomal sites to be bound during de novo MSL-DCC assembly. These data provide the first documented molecular mechanism through which the dosage compensation machinery distinguishes the X chromosome from an autosome. They highlight fundamental principles in the recognition of complex DNA elements by protein that will have a strong impact on many aspects of chromosome biology (Villa, 2016).
Previous work suggested that MSL2 may tether the MSL-DCC to DNA and that an intact CXC domain is required for X-chromosome discrimination. To assess the DNA-binding specificity intrinsic to MSL2 comprehensively, the Drosophila genome was surveyed for MSL2-binding sites in vitro by DNA immunoprecipitation (DIP). Recombinant MSL2 was incubated with sheared genomic DNA (gDNA) purified from male Drosophila S2 cells. MSL2-bound DNA was recovered and sequenced (Villa, 2016).
Considering the lack of X-chromosome binding selectivity seen in previous in vitro studies, it was not expected to find that MSL2 preferentially retrieved DNA from distinct genomic loci, with a notable enrichment of sequences from the X chromosome. On the X chromosome, the MSL2 binding pattern was remarkably similar to the in vivo pattern that marks the positions of high-affinity binding sites (HAS; or chromatin entry sites) of the MSL-DCC. A total of 57 DIP sites coincided with in vivo HAS, although they show different signal intensities. The results were similar if DIP followed by sequencing (DIP-seq) was performed with gDNA extracted from female cells or synthesized in vitro by whole-genome amplification (excluding the contribution of male-specific RNA contaminants or DNA modifications). It therefore appears that recombinant MSL2 has an intrinsic ability to enrich X-chromosomal sequences from complex genomic DNA (Villa, 2016).
Next, the contribution was assessed of the three known MSL2 domains to DNA binding. Deletion of the RING finger domain that mediates MSL2 interaction with MSL1 and contains E3 ligase activity had no obvious effect. Unexpectedly, however, deletion of a region rich in proline and basic amino acid residues (the Pro/Bas domain) that may bind RNA resulted in the complete loss of DNA binding (Villa, 2016).
Upon deletion or mutation of the CXC domain, binding to a subset of sites was much reduced. Statistical analyses revealed 56 regions that specifically required a functional CXC domain for binding. Notably, these 'CXC-dependent' sites displayed a higher enrichment on the X chromosome. A total of 37 sites mapped to MSL2 in vivo peaks (HAS) on the X chromosome, and 2 sites corresponded to rare cases of autosomal sites that show MSL2 enrichment in vivo. The data suggest that MSL2 interacts with DNA via two domains, CXC and Pro/Bas, and that the CXC domain is the major determinant of the selectivity for the X chromosome. While binding-site specificity can be achieved by cooperation between different transcription factors, the finding suggests that cooperation between two different DNA-binding surfaces within this one protein may also refine its overall binding specificity (Villa, 2016).
Sequence analyses within CXC-dependent and CXC-independent binding sites for MSL2 yielded two distinct motifs. Whereas the CXC-independent binding sites shared low-complexity GA repeats, the CXC-dependent peaks centre around a more complex variation of the MSL response element (MRE), with a notable 5' extension. Remarkably, this novel motif can predict in vivo MSL2 binding (HAS) better than the MRE, as its position weight matrix (PWM) is superior in classifying whether MRE hit regions overlap HAS. Applying low thresholds, 2,667 instances of this motif were found throughout the genome, with an approximately twofold enrichment on the X chromosome. Higher-scoring matches to the consensus sequence tend to be more strongly enriched on the X chromosome. For example, the 34 best matches are 9.8-fold enriched on the X chromosome. However, 18 of those instances were not bound in vitro by MSL2 in a CXC-dependent manner, indicating that the recognition sequence represented by a PWM cannot fully explain this binding mode (Villa, 2016).
PWMs model the base readout of DNA sequences with the implicit assumption that each nucleotide at a given position contributes to binding independently of other positions. Physical interactions of neighbouring base pairs, however, alter the structural conformation of the DNA double helix (often referred to as the DNA shape), which may manifest as variations in the minor groove width, roll, helix twist, and propeller twist. Many proteins depend on both base identity and localized helix shape to recognize their binding site. Using a pentamer-based model built from all-atom Monte Carlo simulations of DNA structures, DNA shape parameters were calculated at each base position of the low-stringency motif hits, with 20-base-pair (bp) extensions on either side. To complement these position-centred features regional mono- and dinucleodide frequencies (k-mers) were also calculated in 4-bp windows along the hit sequences. Principal component analysis (PCA) revealed that a combination of DNA shape and k-mer features was able to separate the two classes of sequences: those that were bound in a CXC-dependent manner (CXC-bound) and those that were not bound in a CXC-dependent manner (non-CXC-bound). Sequences in the latter group were either not bound at all (2,502) or were bound independently of the CXC domain. This suggested that at least some of the DNA features might improve binding prediction. Indeed, classification models constructed with the additional feature sets performed much better than a PWM-score model in predicting CXC-dependent binding sites on all motif hits (Villa, 2016).
Guided by the good performance of the classification model using both the PWM-hit-score and k-mer features, mutations were predicted that would convert robust CXC-bound sites to non-CXC-bound sites. The model suggested that the best discriminating residues would localize to the 5' part of the motif and not to the GA-rich region. To test these predictions, the DIP experiment was modified by mixing appropriately diluted DNA oligonucleotides, representing either a native site or its mutated version, into the genomic DNA. The efficiency of DNA retrieval of experimental oligonucleotides and control genomic loci was quantified by quantitative PCR (qPCR). The results confirmed the predictions, leading to the conclusion that the main determinants for CXC-dependent binding reside within the first eight bases at the 5' end of the consensus motif. Notably, this is the part of the motif that diverges most from the MRE (Villa, 2016).
To investigate further the role of MSL2-binding sites in X chromosome dosage compensation, attempts were made to monitor the interactions of MSL2 with HAS in vivo, with minimal contributions from other DCC subunits. Genetic studies had shown that the assembly of a mature MSL-DCC bound to the non-coding roX RNA in male flies is compromised by inactivating the RNA helicase maleless (MLE). Under those circumstances, the remaining MSL2-MSL1 sub-complex is bound to a small subset of HAS. This scenario was recreated in S2 cells by using RNA interference (RNAi) against mle expression, and MSL2 binding was found to be preferentially retained at HAS, corresponding to CXC-dependent binding sites. The 25 HAS that were most resistant to MLE depletion revealed a shared sequence, bearing a strong resemblance to the CXC-dependent motif. By contrast, the 25 sites most sensitive to MLE depletion (bound only by the complete DCC) shared a GA motif similar to the one found in CXC-independent in vitro binding sites. This suggests that under physiological conditions, the MSL2-MSL1 sub-complex directly contacts a subset of HAS in a CXC-dependent manner in the absence of associated protein and RNA subunits (Villa, 2016).
It is possible that these chromosomal interactions represent an intermediate of MSL-DCC assembly. To test this hypothesis, de novo MSL-DCC assembly was initiated in female Kc cells by reducing the expression of the sex-lethal gene Sxl. The SXL protein prevents MSL2 expression and thus the dosage compensation program in female cells. Upon depletion of SXL, binding of newly expressed MSL2 to CXC-dependent HAS was stronger and occurred earlier when compared to CXC-independent ones. Consistent with this finding, hierarchical clustering of MSL2 signals from the common set of Kc and S2 peak regions revealed 30 sites that acquire strong MSL2 binding ability 3 days after SXL depletion. De novo motif discovery on these sites revealed a consensus sequence that resembles the one in the CXC-dependent sites. The data strongly suggest that those sites identified in vitro as CXC-dependent are pioneering binding sites for MSL2 in vivo. These are therefore refered to as PionX sites, and to their defining motif as the PionX motif (Villa, 2016).
The notion that PionX sites are important for dosage compensation is further supported by evolutionary considerations. Drosophila miranda represents a unique system to study how newly evolving X chromosomes acquired dosage compensation. The D. miranda neo-X chromosome is a sex chromosome that began to evolve just 1 million-2 million years ago. Owing to the relatively short evolutionary time span, the neo-X chromosome still retains many autosomal features, but has already acquired partial dosage compensation. Recent work has identified the MSL-DCC-binding sites on all D. miranda X chromosomes19. De novo motif analysis yielded the typical GA-rich MREs for the older, fully compensated X-chromosomal arms XL and XR. Notably, though, the consensus sequence derived from the neo-X chromosome clearly resembled the PionX signature (Villa, 2016).
The neo-Y chromosome originated from the fusion of one Müller-C chromosome to the Y chromosome, resulting in evolutionary pressure on the second Müller-C chromosome to become the neo-X chromosome. The PionX motif (but not the MRE) is particularly enriched on the D. miranda neo-X chromosome but not on the related Drosophlia pseodoobscura Müller C autosome, supporting the idea that this motif represents a new X-chromosome-specific feature. Careful comparison of neo-X-chromosome sequences with the homologous regions in D. pseudoobscura revealed that the novel MSL-DCC-binding sites were acquired by diverse molecular mechanisms, including point mutations and short insertions/deletions of precursor sequences. About half of them originated from precursor sequences contained in a D. miranda-specific helitron transposon. The homologous neo-Y helitron does not contain PionX motifs -- only precursor sequences in which the 5' CAC motif and the 3' GA-rich element are separated. On the neo-X chromosome these two parts are fused by a 10-bp deletion to form PionX consensus motifs. The insertion of a PionX consensus motif derived from the D. miranda neo-X chromosome into an autosome of D. melanogaster led to strong, ectopic binding of the MSL-DCC. By contrast, the corresponding homologous neo-Y-chromosome sequence, in which the 5' and GA sequences are split by a 10-bp insertion, did not recruit the complex. A similar experiment used the strongest DCC-binding site from the neo-X chromosome and the corresponding neo-Y-chromosomal fragment, showing MSL-DCC recruitment to the former, but not to the latter. While the neo-Y-chromosomal fragment does not contain a PionX motif, the evolved neo-X chromosome contains nine of them. Collectively, these observations suggest that PionX motifs play an important role in de novo acquisition of dosage compensation (Villa, 2016).
In summary, this study provides three lines of argument suggesting that PionX sites are X-chromosome-specific determinants that function early in the series of events that lead to exclusive targeting of the X chromosome and correct dosage compensation. First, PionX sites are bound by an MSL2-MSL1 sub-complex in the absence of all other subunits, a state that may reflect an early intermediate of MSL-DCC assembly at HAS. Second, PionX sites are the first to be occupied during de novo establishment of dosage compensation. Finally, PionX motifs arose during the early phase of neo-X-chromosome evolution in D. miranda (Villa, 2016).
A pertinent conceptual advance from this study is the understanding that not all HAS contain the same amount of information. The subset of PionX sites are not necessarily sites of highest MSL2 occupancy in vivo, but contribute an important qualitative element of X-chromosomal discrimination. This discrimination is not wholly apparent from the consensus motif as it also relies on the shape of the DNA at the MSL2-binding site (Villa, 2016).
The initial recruitment of MSL2 to PionX sites on the X chromosome may trigger the distribution of the complex to nearby non-PionX HAS within the chromosomal territory, thereby further amplifying the difference in MSL2 occupancy between the X chromosome and the autosomes. It is likely that other factors contribute to the stability of the targeting system in vivo, such as the cooperativity of MSL2 domains within what is presumed to be a dimeric complex; the assembly of functional complexes within the X-chromosomal territory owing to transcription of roX RNA from the X chromosome; synergistic interactions between different MSL-DCC complexes and with the CLAMP protein at clustered MREs24; and a supportive organization of the conformation of the X chromosome (Villa, 2016).
A common feature of sex chromosomes is coordinated regulation of X-linked genes in one sex. Drosophila melanogaster males have one X chromosome, whereas females have two. The resulting imbalance in gene dosage is corrected by increased expression from the single X chromosome of males, a process known as dosage compensation. In flies, compensation involves recruitment of the male-specific lethal (MSL) complex to X-linked genes and modification of chromatin to increase expression. The extraordinary selectivity of the MSL complex for the X chromosome has never been explained. Previously work has demonstrated that the small interfering RNA (siRNA) pathway and siRNA from a family of X-linked satellite repeats (1.688X repeats) promote X recognition. This study now tests the ability of 1.688X DNA to attract compensation to genes nearby; autosomal integration of 1.688X repeats is shown to increase MSL recruitment and gene expression in surrounding regions. Placement of 1.688X repeats opposite a lethal autosomal deletion achieves partial rescue of males, demonstrating functional compensation of autosomal chromatin. Females block formation of the MSL complex and are not rescued. The 1.688X repeats are therefore cis-acting elements that guide dosage compensation. Furthermore, 1.688X siRNA enhances rescue of males with a lethal deletion but only when repeat DNA is present on the intact homolog. It is proposed that the siRNA pathway promotes X recognition by enhancing the ability of 1.688X DNA to attract compensation in cis. The dense and near-exclusive distribution of 1.688X sequences along the X chromosome suggests that they play a primary role in determining X identity during dosage compensation (Joshi, 2017).
X chromosome dosage compensation in Drosophila requires chromosome-wide coordination of gene activation. The male-specific lethal dosage compensation complex (DCC) identifies and binds to X-chromosomal high-affinity sites (HAS) from which it boosts transcription. A sub-class of HAS, PionX sites, represent first contacts on the X. This study explored the chromosomal interactions of representative PionX sites by high-resolution 4C and determined the global chromosome conformation by Hi-C in sex-sorted embryos. Male and female X chromosomes display similar nuclear architecture, concordant with clustered, constitutively active genes. PionX sites, like HAS, are evenly distributed in the active compartment and engage in short- and long-range interactions beyond compartment boundaries. Long-range, inter-domain interactions between DCC binding sites are stronger in males, suggesting that the complex refines chromatin organization. By de novo induction of DCC in female cells, the extent of activation surrounding PionX sites was monitored. This revealed a remarkable range of DCC action not only in linear proximity, but also at megabase distance if close in space, suggesting that DCC profits from pre-existing chromosome folding to activate genes (Schauer, 2017).
Transcription regulators select their genomic binding sites from a large pool of similar, non-functional sequences. Although general principles that allow such discrimination are known, the complexity of DNA elements often precludes a prediction of functional sites. The process of dosage compensation in Drosophila allows exploring the rules underlying binding site selectivity. The male-specific-lethal (MSL) Dosage Compensation Complex (DCC) selectively binds to some 300 X chromosomal 'High Affinity Sites' (HAS) containing GA-rich 'MSL recognition elements' (MREs), but disregards thousands of other MRE sequences in the genome. The DNA-binding subunit MSL2 alone identifies a subset of MREs, but fails to recognize most MREs within HAS. The 'Chromatin-linked adaptor for MSL proteins' (CLAMP) also interacts with many MREs genome-wide and promotes DCC binding to HAS. Using genome-wide DNA-immunoprecipitation extensive cooperativity id described between both factors, depending on the nature of the binding sites. These are explained by physical interaction between MSL2 and CLAMP. In vivo, both factors cooperate to compete with nucleosome formation at HAS. The male-specific MSL2 thus synergises with a ubiquitous GA-repeat binding protein for refined X/autosome discrimination (Albig, 2018).
Acetylation of histone H4 at lysine 16 (H4K16) modulates nucleosome-nucleosome interactions and directly affects nucleosome binding by certain proteins. In Drosophila, H4K16 acetylation by the dosage compensation complex subunit Mof is linked to increased transcription of genes on the single X chromosome in males. This study analyzed Drosophila containing different H4K16 mutations or lacking Mof protein. An H4K16A mutation causes embryonic lethality in both sexes, whereas an H4K16R mutation permits females to develop into adults but causes lethality in males. The acetyl-mimic mutation H4K16Q permits both females and males to develop into adults. Complementary analyses reveal that males lacking maternally deposited and zygotically expressed Mof protein arrest development during gastrulation, whereas females of the same genotype develop into adults. Together, this demonstrates the causative role of H4K16 acetylation by Mof for dosage compensation in Drosophila and uncovers a previously unrecognized requirement for this process already during the onset of zygotic gene transcription (Copur, 2018).
Mutational analyses of histone amino acid residues that are subject to posttranslational modifications provide a direct approach for probing the physiological role of these residues and their modification. This study investigated the function of H4K16 and its acetylation in Drosophila by generating animals in which all nucleosomes in their chromatin were altered to constitutively carry a positively charged H4R16, an acetyl-mimic H4Q16, or a short apolar H4A16 substitution. These three types of chromatin changes have different physiological consequences that lead to the following main conclusions. First, H4R16 and H4Q16 chromatin both support development of female zygotes into adults. This suggests that, in females, modulation of H4K16 by acetylation is a priori not essential for the regulation of gene expression and the chromatin folding that occurs during development of the zygote. Second, unlike in females, only H4Q16 but not H4R16 chromatin supports development of male embryos into adults. This difference between males and females directly supports the critical role of H4K16 acetylation for dosage compensation in males. Third, cells with H4A16 chromatin are viable, proliferate, and can differentiate to form normal tissues in both males and females, but animals that entirely consist of cells with H4A16 chromatin arrest development at the end of embryogenesis. This lethality contrasts with the viability of animals with H4K16, H4R16, or H4Q16 chromatin and suggests that presence of a long aliphatic side chain with a polar group (i.e., either K, R, or Q) at residue 16 is more important for H4 function than the ability to regulate the charge of this residue by acetylation. A fourth main conclusion of this work comes from the finding that males that completely lack Mof protein (i.e., mof m-z- males) arrest development during gastrulation, whereas females of the same genotype develop into morphologically normal adults. This uncovers a previously unknown critical requirement of Mof acetyltransferase activity in males, already during the onset of zygotic gene transcription. The following sections discuss the results reported in this study in the context of the current understanding of the role of H4K16 and its acetylation (Copur, 2018).
In yeast and flies, the comparison of the severities of the phenotypes caused by different amino acid substitutions at H4K16 highlights how the two organisms have evolved to use this conserved residue and its modification in different ways. In yeast, H4K16ac is present genome-wide and SIR silencing is the key physiological process that requires H4K16, in its deacetylated state. Yeast cells with H4K16R, H4K16Q, or H4K16A mutations are viable but they show defective SIR silencing. Silencing is much more strongly impaired in H4K16Q or H4K16A mutants than in H4K16R mutants. This is because SIR3 protein binding to deacetylated H4K16, a prerequisite for silencing, is probably less severely impaired by the arginine substitution than by the alanine or glutamine substitutions. In Drosophila, the phenotypic differences between H4K16R, H4K16Q, and H4K16A mutants suggest that H4K16 is associated with two other, distinct physiological functions that are critical for the organism. The male-specific lethality of H4K16R mutants and the restoration of male viability in H4K16Q mutants demonstrate that dosage compensation is one essential process that critically requires the acetylated form of H4K16. A reduction of internucleosomal contacts by H4K16ac to generate chromatin that is more conducive to gene transcription on the male X chromosome currently is the simplest mechanistic explanation for how H4K16 acetylation enables dosage compensation. The observation that an H4K16A mutation causes lethality in both sexes suggests that, unlike in yeast, a long aliphatic side chain at this residue is essential for H4 function in Drosophila. It is currently not known why Drosophila H4K16A mutants die. However, it is important to note that H4K16A mutant cells retain the capacity to proliferate and differentiate and the mutation therefore does not disrupt any fundamental process required for cell survival (Copur, 2018).
Previous studies that investigated the function of histone H3 modifications by histone replacement genetics showed that for modifications associated with transcriptionally active chromatin it is essential to remove not only the wild-type copies of the canonical histone genes but to also mutate the histone H3.3 variants. The analyses of H4K16R, H4K16Q, and H4K16A mutant phenotypes reported in this study were all performed in the genetic background of animals lacking His4r, the only histone H4 variant in Drosophila. Importantly, it was found that in a His4r+ background, where only the canonical H4 proteins are replaced with mutant H4, the modifiable His4r protein permitted H4K16R His4r+ mutant males and, surprisingly, also H4K16A His4r+ mutant females and males to develop into adults. These animals were therefore not analyzed further. Supporting these observations, a recent study that used a similar strategy for replacing canonical histone H4 with H4K16R also found that H4K16R His4r+ mutant males develop into normal adults. This suggests that, like His3.3, the His4r protein might also preferentially be incorporated into transcriptionally active chromatin and become acetylated by Mof. Although the viable H4K16R His4r+ males have been reported to show a significant reduction of X-linked gene expression, a full assessment of transcriptional defects in animals containing only H4R16 nucleosomes would require that such molecular analyses be performed in H4K16R His4rΔ mutant males (Copur, 2018).
A final point that should be noted here is that during the early stages of embryogenesis, H4K16R, H4K16Q, or H4K16A mutants also still contain maternally deposited wild-type H4 protein that becomes incorporated into chromatin during the preblastoderm mitoses and only eventually becomes fully replaced by mutant H4 proteins during postblastoderm cell divisions. During the earliest stages of embryogenesis it has therefore not been possible to assess the phenotype of animals with chromatin containing exclusively H4R16, H4Q16, or H4A16 nucleosomes. This needs to be kept in mind when considering comparisons between the phenotypes of H4K16 point mutants and mof m-z- mutants (Copur, 2018).
Males without Mof protein (i.e., mof m-z- males) arrest development during gastrulation while their female siblings develop into adults. Moreover, mof m-z+ males also fail to develop, demonstrating that zygotic expression of Mof protein is insufficient to rescue male embryos that lacked maternally deposited Mof protein. The most straightforward explanation for these observations is that H4K16 acetylation by Mof is critically required for hypertranscription of X-chromosomal genes that has been reported to occur already during the onset of zygotic gene transcription and that the early developmental arrest of males is a direct consequence of failed dosage compensation (Copur, 2018).
How does this early requirement for Mof activity at the blastoderm stage relate to current understanding of the temporal requirement for the DCC for dosage compensation? Previous studies showed that males lacking the DCC subunits Msl-1, Msl-2, Msl-3, or Mle complete embryogenesis and arrest development much later, around the stage of puparium formation. For example, Msl-1 protein null mutants (i.e., msl-1 m-z- mutants) die as late third instar larvae, yet Msl-1 directly interacts with Mof to incorporate it into the DCC and is critical for targeting the complex and H4K16ac accumulation on the X chromosome in larvae. One possible explanation for the conundrum that the lack of Mof but not that of Msl-1 or other DCC subunits results in lethality during gastrulation could be that during these early stages, H4K16 acetylation by Mof for dosage compensation is not as strictly dependent on the other DCC subunits as during later developmental stages, or that there is redundancy between Msl-1, Msl-2, or Msl-3 for targeting Mof to the X chromosome in the early embryo (Copur, 2018).
A final point worth noting is that Mof is also present in another protein assembly called the NSL complex. NSL was reported to act genome-wide for regulating housekeeping gene transcription in both sexes and several NSL subunits are essential for Drosophila viability. The finding that mof m-z- mutant females develop into morphologically normal adults shows that the NSL complex must exert regulatory functions that are essential for viability independently of Mof H4K16 acetyltransferase activity (Copur, 2018).
The acetylation of lysine residues in the N termini of histones is generally associated with chromatin that is conducive to gene transcription. Mutational studies in yeast showed that there is substantial functional redundancy between most of the different acetylated lysine residues in the N termini of histone H3 and H4 but that H4K16 has unique effects on transcriptional control, with well-defined phenotypic consequences. This study shows that in Drosophila the principal function of H4K16 acetylation is X-chromosome dosage compensation in males (Copur, 2018).
In Drosophila melanogaster, the male-specific lethal (MSL) complex plays a key role in dosage compensation by stimulating expression of male X-chromosome genes. It consists of MSL proteins and two long noncoding RNAs, roX1 and roX2, that are required for spreading of the complex on the chromosome and are redundant in the sense that loss of either does not affect male viability. However, despite rapid evolution, both roX species are present in diverse Drosophilidae species, raising doubts about their full functional redundancy. Thus, this study investigated consequences of deleting roX1 and/or roX2 to probe their specific roles and redundancies in D. melanogaster. Anew mutant allele of roX2 was created, and roX1 and roX2 were shown to have partly separable functions in dosage compensation. In larvae, roX1 is the most abundant variant and the only variant present in the MSL complex when the complex is transmitted (physically associated with the X-chromosome) in mitosis. Loss of roX1 results in reduced expression of the genes on the X-chromosome, while loss of roX2 leads to MSL-independent upregulation of genes with male-biased testis-specific transcription. In roX1 roX2 mutant, gene expression is strongly reduced in a manner that is not related to proximity to high-affinity sites. These results suggest that high tolerance of mis-expression of the X-chromosome has evolved. It is proposed that this may be a common property of sex-chromosomes, that dosage compensation is a stochastic process and its precision for each individual gene is regulated by the density of high-affinity sites in the locus (Kim, 2018).
The dosage compensation machinery involving rox1 and rox2 RNAs provides a valuable model system for studying the evolution of lncRNA-genome interactions, chromosome-specific targeting and gene redundancy. LncRNAs differ from protein coding genes and are often less conserved at the level of primary sequence, as expected due to their lack of protein-coding restrictions. Like those encoding other lncRNAs, rapid evolution, i.e., low conservation of the primary sequences of roX genes has complicated comparative studies. Despite their differences in length and primary sequences, rox1 and rox2 have also been considered functionally redundant in Drosophila melanogaster. However, remarkably considering their rapid evolution and apparent redundancy, orthologs for both rox1 and rox2 have been found in all of 26 species within the Drosophila genus with available whole genome assemblies. Models that explain evolutionarily stable redundancy have been proposed suggesting that the presence of both rox1 and rox2 in these diverged species may be attributable to differences in targets, affinities and/or efficiency or additional functions (Kim, 2018).
On polytene chromosomes, binding patterns of rox1 and rox2 are more or less indistinguishable, except in region 10C where rox2 is almost exclusively present. In the rox2 mutant, genes located in the 10C bin are on average downregulated, but similar downregulation of genes in many other bins is observed, so the effect cannot be directly attributed to loss of rox2. In wildtype 1st instar larvae, levels of rox1 RNA are much higher than levels of rox2 RNA. Interestingly, in rox1 mutant larvae the absolute amount of rox2 RNA increases, but only to ~10% of wildtype levels of total roX RNA. This appears sufficient to avoid lethality, but still causes a significant decrease in X-chromosome expression. However, despite the huge difference in amounts, not only in number but even more considering the size of the two roX RNAs, the staining intensities of roX RNA on rox1 mutant and wildtype polytene chromosomes seem to be roughly equal. On mitotic chromosomes rox1 RNA is only observed in the MSL complexes bound to the distal X-chromosome and this binding is not redundant. This indicates that just after cell division rox1 RNA will be the dominating variant in assembled MSL complexes. Taken together, the results suggest that rox2 RNA has higher affinity than rox1 RNA for inclusion in MSL complexes. Moreover, varying amounts of the two species with different affinities at given cell cycle stages may support proper transmission, spreading of assembled MSL complexes and maintenance of appropriate levels of the complexes (Kim, 2018).
It should be noted that some male rox1 rox2 mutants escaped, so loss of roX is not completely male-lethal, unlike loss of mle, msl1, msl2, msl3 or mof. The complete male lethality in these mutants is attributed to reductions in dosage compensation that have been measured in several studies and observed not only in msl mutants but also following RNAi-mediated depletion of MSL proteins. Notably, the average reduction of X-chromosome expression, relative to wildtype levels, calculated in these cases has varied from ca. 20 to 30%; substantially less than the 35% reduction observed in the rox1 rox2 mutant. Some of the reported differences may be due to use of different techniques and bioinformatics procedures (including use of different cut-offs for expression and developmental stages). However, the reasons why some males can survive the very dramatic imbalance observed in expression of a large portion of the genome are unclear. Furthermore, the reduction in expression of X-chromosome genes observed in the rox1 mutant is not accompanied by any reported phenotypic changes, indicating that D. melanogaster has high intrinsic ability to cope with significant imbalances in X-chromosome expression. It is speculated that in parallel with a compensation mechanism that addresses dosage imbalances the fly has evolved a high degree of tolerance to mis-expression of the X-chromosome (Kim, 2018).
Evolutionary studies have shown that sex chromosomes do not always represent terminal stages in evolution-in fact, the 4th chromosome was ancestrally an X-chromosome that reverted to an autosome. Moreover, the fly shows high and unusual tolerance to dosage differences and mis-expression of the 4th chromosome (although much smaller than the tolerance to those of the X-chromosome). These observations suggest that tolerance of mis-expression is a common outcome in the evolution of sex-chromosomes and this property has been retained with respect to the 4th chromosome, even after its reversion to an autosome. It is proposed that high tolerance of mis-expression in the absence of full functional dosage compensation may be selected for during evolution of sex-chromosomes. This is because gradual degeneration of the proto-Y chromosome will be accompanied by an increasing requirement to equalize gene expression between a single X- (in males) and two X-chromosomes (in females), but changes in genomic location of highly sensitive genes will be favored during periods of incomplete (or shifting) dosage compensation. On transcript level, responses to reductions in dosages of X-chromosome genes have been found to be similar to those of autosomal genes. Thus, potential mechanisms for the higher tolerance are post-transcriptional compensatory mechanisms or selective alterations in gene composition (changes in genomic locations), similar to those proposed for the observed demasculinization of the Drosophila X-chromosome (Kim, 2018).
Prompted by the strong relationship between orchestration of the X- and 4th chromosomes by the MSL complex and POF system, respectively, this study also measured effects of roX suppression on chromosome 4 expression in roX mutants. Weak but significant reduction of expression were observed in the rox2 mutant, but the cause of this reduction remains elusive. In rox2 mutant transcriptional upregulation of X-chromosome genes classified as having low expression levels was also oberved, late replication and weak MSL complex-binding. The loss of rox2 resulting in MSL complexes only including rox1 RNA might alter the spreading properties. It is therefore hypothesized that the observed upregulation might be caused by mis-targeting of the MSL complex in the absence of rox2. However, the ChIP experiment revealed no enrichment of MSL complexes on these genes, and results rather suggest that rox2 directly or indirectly restricts expression of these male-biased genes independently of its role in the MSL complex (Kim, 2018).
It is well known that roX RNAs are important for spreading of the MSL complex in regions between high affinity sites (HAS). It is therefore surprising that loss of roX causes a relatively even reduction in expression of X-chromosomal genes and the decrease is not more dramatic with larger distances, as would be expected for reductions in spreading capacity. Indeed, observed reductions in expression were smaller for genes located far from HAS than for closer genes. A possible explanation is that expression of these genes is compensated by an MSL-independent mechanism. It has been previously shown that most genes on the X-chromosome are dosage-compensated, but a subset are not bound by the MSL complex and do not respond to its depletion. The results corroborate these findings since loss of roX RNA in the rox1 rox2 mutant had little effect on the expression of genes classified as having weak MSL complex binding, clearly indicating that at least one other mechanism is involved. The results further show that high-affinity sites, as defined by MSL-targets in the absence of rox1 and rox2, are highly correlated to genes with the highest MSL binding levels. Therefore, sites targeted in the absence of roX provide a more stringent definition of HAS, with stronger correlation to genes bound by high levels of MSL complex, than targets in the absence of mle, mof or msl3 (Kim, 2018).
The increase in expression mediated by the MSL complex is considered a feed-forward mode of regulation, and appears to be more or less equal (ca. 35%) for all MSL-bound genes. Evidently, highly expressed genes need a stronger increase in transcription than weakly-expressed genes. These results suggest that dosage compensation is a stochastic process that depends on HAS distribution and is correlated with expression levels. Evolutionary analysis has shown that newly formed X-chromosomes acquire HAS, putatively via rewiring of the MSL complex by transposable elements and fine-tuning of its regulatory potential. Such a dynamic process may be required for constant adaptation of the system. Highly expressed genes tend to accumulate HAS in their introns and 3'UTRs, and thus bind relatively high amounts of MSL complex, thereby stimulating the required increase in expression. This also implies that the gene organization on X-chromosomes is under more constraints than autosomes (Kim, 2018).
This study presents the first high-throughput sequencing data and analysis of transcriptomes of rox1, rox2 and rox1 rox2 mutant flies. The results reveal that rox1 and rox2 fulfill separable functions in dosage compensation in D. melanogaster. The two RNA species differ in both transcription level and cell-cycle regulation (Kim, 2018).
In third instar larvae, rox1 is the more abundant variant and the variant that is included in MSL complexes transmitted physically associated with the X-chromosome in mitosis. Loss of rox1, but not loss of rox2, results in decreased expression of genes on the X-chromosome, albeit without apparent phenotypic consequences. Loss of both roX species leads to a dramatic reduction of X-chromosome expression, but not complete male lethality. Taken together, these findings suggest that high tolerance for mis-expression of X-chromosome genes has evolved. It is speculated that it evolved in parallel with dosage compensation mechanisms and that it may be a common property of current and ancient sex-chromosomes (Kim, 2018).
The roX RNAs are important for spreading of the MSL-complex from HAS, but the reduction of X-chromosome expression in rox1 rox2 mutant is not affected by the need for spreading, i.e., distance from HAS. In addition, the genes targeted by the MSL complex in the rox1 rox2 mutant also show strongly reduced expression. The results suggest that the function of the MSL complex which is still present at HAS is compromised in the rox1 rox2 mutant and that the dosage of distant genes is compensated by an alternative, unknown, mechanism. It is proposed that dosage compensation is a stochastic process that depends on HAS distribution. Creation and fine-tuning of binding sites is a dynamic process that is required for constant adaptation of the system. Highly expressed genes will accumulate and be selected for strong HAS (and thus bind more MSL complex) since they require high levels of bound MSL complex for the required increases in expression (Kim, 2018).
The repeatability or predictability of evolution is a central question in evolutionary biology and most often addressed in experimental evolution studies. This study inferred how genetically heterogeneous natural systems acquire the same molecular changes to address how genomic background affects adaptation in natural populations. In particular, advantage was taken of independently formed neo-sex chromosomes in Drosophila species that have evolved dosage compensation by co-opting the dosage-compensation male-specific lethal (MSL) complex to study the mutational paths that have led to the acquisition of hundreds of novel binding sites for the MSL complex in different species. This complex recognizes a conserved 21-bp GA-rich sequence motif that is enriched on the X chromosome, and newly formed X chromosomes recruit the MSL complex by de novo acquisition of this binding motif. Recently formed sex chromosomes were identified in the D. melanica and D. robusta species groups by genome sequencing and generate genomic occupancy maps of the MSL complex to infer the location of novel binding sites. Diverse mutational paths were utilized in each species to evolve hundreds of de novo binding motifs along the neo-X, including expansions of microsatellites and transposable element (TE) insertions. However, the propensity to utilize a particular mutational path differs between independently formed X chromosomes and appears to be contingent on genomic properties of that species, such as simple repeat or TE density. This establishes the 'genomic environment' as an important determinant in predicting the outcome of evolutionary adaptations (Ellison, 2019).
This study took advantage of naturally occurring variation in sex chromosome karyotype in Drosophila species to study independent replicates of solving the same evolutionary challenge: to dosage compensate newly formed neo-X chromosomes by acquiring hundreds of MSL-binding sites in response to Y degeneration (Ellison, 2019).
The independent acquisition of dosage compensation in Drosophila allows several important questions in evolutionary biology and gene regulation to be addressed: first, how repeatable is evolution? Evolutionary biologists have long debated the predictability of the evolutionary process. At one extreme, evolution could be highly idiosyncratic and unpredictable, since the survival of the fittest could occur along a great number of forking paths. Alternatively, constraints on evolution may force independent lineages to frequently converge on the same genetic solutions for the same evolutionary challenge. Second, how do regulatory networks evolve? And what is the contribution of TEs to regulatory evolution? Evolutionary innovations and adaptations often require rapid and concerted changes in regulation of gene expression at many loci. TEs constitute the most dynamic part of eukaryotic genomes, and the dispersal of TEs that contain a regulatory element may allow for the same regulatory motif to be recruited at many genomic locations, thereby drawing multiple genes into the same regulatory network. Third, what makes a binding motif functional? The genomes of complex organisms encompass megabases of DNA, and regulatory molecules must distinguish specific targets within this vast landscape. Regulatory factors typically identify their targets through sequence-specific interactions with the underlying DNA, but they typically bind only a fraction of the candidate genomic regions containing their specific target sequence motif. An unresolved mystery in regulatory evolution is what drives the specificity of binding to a subset of genomic regions that all appear to have a sequence that matches the consensus binding motif (Ellison, 2019).
Several features make dosage compensation in Drosophila a promising system to tackle these questions. The genetic architecture for most adaptations -- especially those involving regulatory changes -- as well as the timing and exact selective forces driving them is generally little understood. In contrast, detailed knowledge is available of the molecular mechanism of dosage compensation in Drosophila. The cis- and trans-acting components of this regulatory network and the regulatory motif for targeting the MSL complex to the X are known. Clear expectations are available of which genomic regions should acquire dosage compensation and about the timing and the evolutionary forces that drive wiring of hundreds of genes into the dosage-compensation network on newly evolved X chromosomes. Specifically, Y degeneration is a general facet of sex chromosome evolution, creating selective pressures to up-regulate X-linked genes in males. Dosage compensation should thus only evolve on neo-X chromosomes whose neo-Y homologs have started to degenerate and should evolve simultaneously or shortly after substantial gene loss has occurred on the neo-Y. Indeed, comparative data in Drosophila support this model of dosage-compensation evolution. Drosophila species with partially eroded neo-Y chromosomes exist that have not yet evolved MSL-mediated dosage compensation, including D. busckii and D. albomicans, lending empirical support to the notion that dosage compensation evolves in response to Y degeneration and not the other way round. Thus, a refined understanding of how, when, why, and where dosage compensation in Drosophila evolves makes this an ideal model system to study the repeatability of evolution and the evolution of regulatory networks (Ellison, 2019).
Results from evolution experiments indicate that although evolution is not identical in replicate populations, there is an important degree of predictability. Experimentally evolved populations under controlled, identical conditions consistently show parallelism in which mutations in certain genes are repeatedly selected. However, organisms adapting to similar environments are not genetically identical, but their genome instead carries the legacy of their unique evolutionary trajectory, raising the question of how genomic differences affect genetic parallelism (Ellison, 2019).
Sex chromosome-autosome fusions have independently created neo-sex chromosomes in different Drosophila lineages. This provides everal independent replicates to study how, on the molecular level, evolution has solved the same adaptive challenge: acquiring hundreds of binding sites to recruit the MSL complex to newly formed X chromosomes. This allows quantification of how much variation there is, both within and between species, in the underlying mutational paths to acquire hundreds of MSL-binding sites on neo-X chromosomes and identify genomic contingencies that will influence the repeatability of evolutionary trajectories. Importantly, neo-sex chromosomes of Drosophila are evolutionarily young (between 0.1-15 MY old), which allows, in many cases, inferring of the causative mutations that have resulted in the gain of a regulatory element and decipher the evolutionary processes at work to draw hundreds of genes into a new regulatory network (Ellison, 2019).
The results suggest that the evolution of MSL-binding sites is highly opportunistic but contingent on genomic background. In particular, each independently evolved neo-X chromosome was found to use a diverse set of mutational pathways to acquire MSL-binding sites on a new neo-X chromosome, ranging from microsatellite expansions to the utilization of presites to TE insertions. However, different lineages differ with regards to the frequency of which mutational paths are most often followed to acquire novel binding sites, and this propensity may depend on the genomic background. In particular, species found with the higher density of simple repeats are more prone to utilize expansions in GA microsatellites to gain a novel MSL-binding site. In contrast, D. robusta has an elevated TE density compared to its sibling species, and it was found that the dispersal of a TE has played an important role in the acquisition of MSL-binding sites on its neo-X chromosome. Thus, this suggests that the genomic background of a species predisposes it to evolve along a particular path, yet the evolutionary process is random and resourceful with regards to utilizing a variety of mutations to create novel MSL-binding sites. However, different phenotypes show drastic differences in their underlying genetic architecture, and the importance of genomic background likely differs among traits and (Ellison, 2019).
Evolutionary innovations and adaptations often require rapid and concerted changes in regulation of gene expression at many loci. It has been suggested that TEs play a key role in rewiring regulatory networks, since the dispersal of TEs that contain a regulatory element may allow for the same regulatory motif to be recruited at many genomic locations. A handful of recent studies have implicated TEs as drivers of key evolutionary innovations, including placentation in mammals or rewiring the core regulatory network of human embryonic stem cells. While these studies demonstrate that TEs can, in principle, contribute to the creation or rewiring of regulatory networks, they do not address the question of how often regulatory elements evolve by TE insertions versus by other mutations. That is, the importance of TEs in contributing to regulatory evolution is not known. Quantification of the role of TEs would require a priori knowledge of how and when regulatory networks evolve and a detailed molecular understanding of which genes are being drawn into a regulatory network and how. As discussed above, these parameters are well understood for dosage compensation in flies (Ellison, 2019).
Previous work in D. miranda has shown that a helitron TE was recruited into the dosage-compensation network at two independent time points. The younger 1.5-MY-old neo-X chromosome of D. miranda is in the process of evolving dosage compensation, and dozens of new CESs on this chromosome were created by insertions of the ISX element. The domesticated ISX TE gained a novel MRE motif by a 10-bp deletion in the ISY element, which is a highly abundant TE in the D. miranda genome. The remnants of a related (but different) TE at CES was found on the older neo-X of this species (which formed roughly 13-15 MY ago), but the TE was too eroded to reconstruct its evolutionary history. This study, identified another domesticated TE that was utilized to deliver MSL-binding sites to a newly formed neo-X chromosome, but no significant TE contribution was found for MSL-binding site evolution in two independent neo-X chromosomes (Ellison, 2019).
The data shed light on the question of when TEs are expected to be important in regulatory evolution. For TEs to contribute to regulatory rewiring, two conditions have to be met: a regulatory element (or a progenitor sequence that can easily mutate into the required binding motif) needs to be present in the TE, and TE needs to be active in the genome (and not yet silenced by the host machinery). TEs undergo a characteristic life cycle in which they invade a new species (or escape the genome defense by mutation) and transpose until they are silenced by the host genome. Once a TE is robustly repressed, it no longer can serve as a vehicle to disperse regulatory elements, so the time window when a particular TE family can be domesticated is probably short and needs to coincide with a necessity to disperse regulatory motifs. A high TE burden does increase that chance, but at a cost: maintaining active TEs in the genome allows a rapid response to evolutionary challenges but also creates a major source of genomic mutation, illegitimate recombination, genomic rearrangements, and genome size inflation (Ellison, 2019).
The current findings support this view of a TE tradeoff. The ISY element in D. miranda is the most highly abundant transposon in the D. miranda genome and is massively contributing to the degeneration of the neo-Y in this specie. Indeed, the genomic analysis has revealed >20,000 novel insertions of the ISY element on the neo-Y, often within genes. Yet, it contained a sequence that was only one mutational step away from a functional MSL-binding site (that is, a single 10-bp deletion), and domestication of this element allowed for the rapid dispersal of functional binding sites for the MSL complex along the neo-X. The domestication of the TE in D. robusta occurred too long ago for to reconstruct its exact evolutionary history and the potential damage its mobilization may have caused while it was active. However, consistent with a tradeoff that the host genome faces, it was found that D. robusta has a higher TE density than its sister species and also a considerably larger genome size, yet a TE contributed to wiring hundreds of genes into the dosage-compensation network on its neo-X (Ellison, 2019).
Perhaps surprisingly, in many instances, it was not possible to detect specific mutations that would generate a novel MSL binding motif. Instead, it was found that functional MSL-binding sites are derived from presites containing the GA-rich motif that was already present in an ancestor in which the neo-X is autosomal and in which these sequences do not recruit the MSL complex. The MSL binding motif is only modestly enriched on the X chromosome compared to the autosomes (only approximately 2-fold), and only a small fraction of putative binding sites are actually bound by the MSL complex. The dosage-compensation machinery shares this characteristic with many other sequence-specific binding factors whose predicted target motifs are often in vast excess to the sites actually utilized. It has been speculated that other genomic aspects, such as chromatin context or the 3D organization of the genome, could help to distinguish between utilized and nonutilized copies of a motif. The finding that a large number of sites can acquire the ability to recruit the MSL complex, without any apparent associated changes at the DNA level, supports the view that epigenetic modifications or changes to the 3D architecture of the genome help to ultimately determine which putative binding sites in the genome are actually utilized. In D. melanogaster, the X chromosome has a unique satellite DNA composition, and it was suggested that these repeats play a primary role in determining X identity during dosage compensation. Furthermore, localization of the MSL complex to MREs is dependent on an additional cofactor, the CLAMP protein. CLAMP binds directly to GA-rich MRE sequences and targets MSL to the X chromosome but also binds to GA-rich sequence elements throughout the genome. Recent work has shown that variability in sequence composition within similar GA-rich motifs drive specificity for CLAMP binding, and variation within seemingly similar cis elements may also drive context-specific targeting of the MSL complex. Future investigations of changes in the chromatin level, the repeat content, and the genomic architecture of these newly formed sex chromosomes will help to resolve this outstanding question (Ellison, 2019).
Many heterogametic organisms adjust sex chromosome expression to accommodate differences in gene dosage. This requires selective recruitment of regulatory factors to the modulated chromosome. How these factors are localized to a chromosome with requisite accuracy is poorly understood. Drosophila melanogaster males increase expression from their single X chromosome. Identification of this chromosome involves cooperation between different classes of X-identity elements. The Chromatin Entry Sites (CES) recruit a chromatin-modifying complex that spreads into nearby genes and increases expression. In addition, a family of satellite repeats that is enriched on the X chromosome, the 1.688(X) repeats, promotes recruitment of the complex to nearby genes. The 1.688(X) repeats and CES are dissimilar, and appear to operate through different mechanisms. Interestingly, the siRNA pathway and siRNA from a 1.688(X) repeat also promote X recognition. This study postulated that siRNA-dependent modification of 1.688(X) chromatin contributes to recognition of nearby genes. In accord with this, enrichment of the siRNA effector Argonaute2 (Ago2) was found at some 1.688(X) repeats. Mutations in several proteins that physically interact with Ago2, including the histone methyltransferase Su(var)3-9, enhance the lethality of males with defective X recognition. Su(var)3-9 deposits H3K9me2 on some 1.688(X) repeats, and this mark is disrupted upon ectopic expression of 1.688(X) siRNA. Furthermore, integration of 1.688(X) DNA on an autosome induces local H3K9me2 deposition, but enhances expression of nearby genes in a siRNA-dependent manner. These findings are consistent with a model in which siRNA-directed modification of 1.688(X) chromatin contributes to recognition of the male X chromosome for dosage compensation (Deshpande, 2018).
Albig, C., Tikhonova, E., Krause, S., Maksimenko, O., Regnard, C. and Becker, P. B. (2018). Factor cooperation for chromosome discrimination in Drosophila. Nucleic Acids Res. PubMed ID: 30541149
Alekseyenko, A. A., Ellison, C. E., Gorchakov, A. A., Zhou, Q., Kaiser, V. B., Toda, N., Walton, Z., Peng, S., Park, P. J., Bachtrog, D. and Kuroda, M. I. (2013). Conservation and de novo acquisition of dosage compensation on newly evolved sex chromosomes in Drosophila. Genes Dev 27: 853-858. PubMed ID: 23630075
Aleman, J. R., Kuhn, T. M., Pascual-Garcia, P., Gospocic, J., Lan, Y., Bonasio, R., Little, S. C. and Capelson, M. (2021). Correct dosage of X chromosome transcription is controlled by a nuclear pore component. Cell Rep 35(11): 109236. PubMed ID: 34133927
Anderson, J. T., Henikoff, S., Ahmad, K. (2023). Chromosome-specific maturation of the epigenome in the Drosophila male germline. Elife, 12 PubMed ID: 38032818
Bachtrog, D., Toda, N. R. and Lockton, S. (2010). Dosage compensation and demasculinization of X chromosomes in Drosophila. Curr. Biol. 20(16): 1476-81. PubMed Citation: 20705467
Beadle, L. F., Zhou, H., Rattray, M. and Ashe, H. L. (2023). Modulation of transcription burst amplitude underpins dosage compensation in the Drosophila embryo. Cell Rep 42(4): 112382. PubMed ID: 37060568
Belyi, A., Argyridou, E. and Parsch, J. (2020). The influence of chromosomal environment on X-linked gene expression in Drosophila melanogaster. Nature. PubMed ID: 33208948
Bhaskar, P. K., Southard, S., Baxter, K. and Van Doren, M. (2022). Germline sex determination regulates sex-specific signaling between germline stem cells and their niche. Cell Rep 39(1): 110620. PubMed ID: 35385723
Billeter, J. C., Atallah, J., Krupp, J. J., Millar, J. G. and Levine, J. D. (2009). Specialized cells tag sexual and species identity in Drosophila melanogaster. Nature 461(7266): 987-91. PubMed Citation: 19829381
Casper, A. L. and Van Doren, M. (2009). The establishment of sexual identity in the Drosophila germline. Development 136(22): 3821-30. PubMed Citation: 19855024
Cheng, B., Kuppanda, N., Aldrich, J.C., Akbari, O.S. and Ferree, P.M. (2016). Male-killing spiroplasma alters behavior of the dosage compensation complex during Drosophila melanogaster embryogenesis. Curr Biol [Epub ahead of print]. PubMed ID: 27161498
Copur, O., Gorchakov, A., Finkl, K., Kuroda, M. I. and Muller, J.
(2018). Sex-specific phenotypes of histone H4 point mutants establish
dosage compensation as the critical function of H4K16 acetylation in
Drosophila. Proc Natl Acad Sci U S A. PubMed ID: 30530664
DeFalco, T., Camara, N., Le Bras, S. and Van Doren, M. (2008). Nonautonomous sex determination controls sexually dimorphic development of the Drosophila gonad. Dev. Cell 14(2): 275-86. PubMed Citation: 18267095
Deshpande, N. and Meller, V. H. (2018). Chromatin that guides dosage compensation is modulated by the siRNA pathway in Drosophila melanogaster. Genetics. PubMed ID: 29921620
Eggers, N., Gkountromichos, F., Krause, S., Campos-Sparr, A. and Becker, P. B. (2023). Physical interaction between MSL2 and CLAMP assures direct cooperativity and prevents competition at composite binding sites. Nucleic Acids Res. PubMed ID: 37602401
Ellison, C. E. and Bachtrog, D. (2013). Dosage compensation via transposable element mediated rewiring of a regulatory network. Science 342: 846-850. PubMed ID: 24233721
Ellison, C. and Bachtrog, D. (2019). Contingency in the convergent evolution of a regulatory network: Dosage compensation in Drosophila. PLoS Biol 17(2): e3000094. PubMed ID: 30742611
Ferrari, F., Plachetka, A., Alekseyenko, A. A., Jung, Y. L., Ozsolak, F., Kharchenko, P. V., Park, P. J. and Kuroda, M. I. (2013). 'Jump Start and Gain' model for dosage compensation in Drosophila based on direct sequencing of nascent transcripts. Cell Rep 5: 629-636. PubMed ID: 24183666
Grmai, L., Hudry, B., Miguel-Aliaga, I. and Bach, E. A. (2018). Chinmo prevents transformer alternative splicing to maintain male sex identity. PLoS Genet 14(2): e1007203. PubMed ID: 29389999
Flaherty, M. S., Salis, P., Evans, C. J., Ekas, L. A., Marouf, A., Zavadil, J., Banerjee, U. and Bach, E. A. (2010). chinmo is a functional effector of the JAK/STAT pathway that regulates eye development, tumor formation, and stem cell self-renewal in Drosophila. Dev Cell 18(4): 556-568. PubMed ID: 20412771
Graze, R. M., Tzeng, R. Y., Howard, T. S. and Arbeitman, M. N. (2018). Perturbation of IIS/TOR signaling alters the landscape of sex-differential gene expression in Drosophila. BMC Genomics 19(1): 893. PubMed ID: 30526477
Gu, L., Reilly, P. F., Lewis, J. J., Reed, R. D., Andolfatto, P. and Walters, J. R. (2019). Dichotomy of Dosage Compensation along the Neo Z Chromosome of the Monarch Butterfly. Curr Biol 29(23): 4071-4077. PubMed ID: 31735674
Guo, J., Tang, H. W., Li, J., Perrimon, N. and Yan, D. (2018). Xio is a component of the Drosophila sex determination pathway and RNA N(6)-methyladenosine methyltransferase complex. Proc Natl Acad Sci U S A 115(14): 3674-3679. PubMed ID: 29555755
Haag, E. S. and Doty, A. V. (2005). Sex determination across
evolution: connecting the dots. PLoS Biol. 3(1): e21. 15660158
Harumoto, T., Fukatsu, T. and Lemaitre, B. (2018a). Common and unique strategies of male killing evolved in two distinct Drosophila symbionts. Proc Biol Sci 285(1875). PubMed ID: 29563258
Harumoto, T. and Lemaitre, B. (2018b). Male-killing toxin in a bacterial symbiont of Drosophila. Nature. PubMed ID: 29720654
Hashiyama, K., Hayashi, Y. and Kobayashi, S. (2011). Drosophila Sex lethal gene initiates female development in germline progenitors. Science 333(6044): 885-8. PubMed Citation: 21737698
Haussmann, I. U., Bodi, Z., Sanchez-Moran, E., Mongan, N. P., Archer, N., Fray, R. G. and Soller, M. (2016). m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540(7632): 301-304. PubMed ID: 27919081
Ilyin, A. A., Kononkova, A. D., Golova, A. V., Shloma, V. V., Olenkina, O. M., Nenasheva, V. V., Abramov, Y. A., Kotov, A. A., Maksimov, D. A., Laktionov, P. P., Pindyurin, A. V., Galitsyna, A. A., Ulianov, S. V., Khrameeva, E. E., Gelfand, M. S., Belyakin, S. N., Razin, S. V. and Shevelyov, Y. Y. (2022). Comparison of genome architecture at two stages of male germline cell differentiation in Drosophila. Nucleic Acids Res 50(6): 3203-3225. PubMed ID: 35166842
Johansson, A. M., Stenberg, P., Allgardsson, A. and Larsson, J. (2012). POF regulates the expression of genes on the fourth chromosome in Drosophila melanogaster by binding to nascent RNA. Mol Cell Biol 32: 2121-2134. PubMed ID: 22473994
Jolma A., Yin Y., Nitta K.R., Dave K., Popov A., Taipale M., Enge M., Kivioja T., Morgunova E., Taipale J. (2015), DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527:384-388 PubMed ID: 26550823
Jordan, W. and Larschan, E. (2021). The zinc finger protein CLAMP promotes long-range chromatin interactions that mediate dosage compensation of the Drosophila male X-chromosome. Epigenetics Chromatin 14(1): 29. PubMed ID: 34187599
Joshi, S. S. and Meller, V. H. (2017). Satellite repeats identify X chromatin for dosage compensation in Drosophila melanogaster males. Curr Biol 27(10): 1393-1402.e1392. PubMed ID: 28457869
Kim, M., Faucillion, M. L. and Larsson, J. (2018). RNA-on-X 1 and 2 in Drosophila melanogaster fulfill separate functions in dosage compensation. PLoS Genet 14(12): e1007842. PubMed ID: 30532158
Kitadate, Y., Shigenobu, S., Arita, K. and Kobayashi, S. (2007). Boss/Sev signaling from germline to soma restricts germline-stem-cell-niche formation in the anterior region of Drosophila male gonads. Dev. Cell 13: 151-159. PubMed Citation: 17609117
Koya, S. K. and Meller, V. H. (2015). Modulation of heterochromatin by male specific lethal proteins and roX RNA in Drosophila melanogaster males. PLoS One 10: e0140259. PubMed ID: 26468879
Krzywinska, E., Ferretti, L., Li, J., Li, J. C., Chen, C. H. and Krzywinski, J. (2021). femaleless Controls Sex Determination and Dosage Compensation Pathways in Females of Anopheles Mosquitoes. Curr Biol. PubMed ID: 33417880
Krzywinska, E., Ribeca, P., Ferretti, L., Hammond, A., Krzywinski, J. (2023). A novel factor modulating X chromosome dosage compensation in Anopheles. Curr Biol, 33(21):4697-4703. PubMed ID: 37774706
Kuzu, G., et al. (2016). Expansion of GA dinucleotide repeats increases the density of CLAMP binding sites on the X-Chromosome to promote Drosophila dosage compensation. PLoS Genet 12: e1006120. PubMed ID: 27414415
Landeen, E. L., Muirhead, C. A., Wright, L., Meiklejohn, C. D. and Presgraves, D. C. (2016). Sex chromosome-wide transcriptional suppression and compensatory cis-regulatory evolution mediate gene expression in the Drosophila male germline. PLoS Biol 14: e1002499. PubMed ID: 27404402
Larschan, E., Bishop, E. P., Kharchenko, P. V., Core, L. J., Lis, J. T., Park, P. J. and Kuroda, M. I. (2011). X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471: 115-118. PubMed ID: 21368835
Lee, H., Cho, D.Y., Whitworth, C., Eisman, R., Phelps, M., Roote, J., Kaufman, T., Cook, K., Russell, S., Przytycka, T. and Oliver, B. (2016). Effects of gene dose, chromatin, and network topology on expression in Drosophila melanogaster. PLoS Genet 12: e1006295. PubMed ID: 27599372 Lence, T., Akhtar, J., Bayer, M., Schmid, K., Spindler, L., Ho, C. H., Kreim, N., Andrade-Navarro, M. A., Poeck, B., Helm, M. and Roignant, J. Y. (2016). m6A modulates neuronal functions and sex determination in Drosophila. Nature 540(7632): 242-247. PubMed ID: 27919077
Mahadevaraju, S., Fear, J. M., Akeju, M., Galletta, B. J., Pinheiro, M., Avelino, C. C., Cabral-de-Mello, D. C., Conlon, K., Dell'Orso, S., Demere, Z., Mansuria, K., Mendonca, C. A., Palacios-Gimenez, O. M., Ross, E., Savery, M., Yu, K., Smith, H. E., Sartorelli, V., Yang, H., Rusan, N. M., Vibranovski, M. D., Matunis, E. and Oliver, B. (2021). Dynamic sex chromosome expression in Drosophila male germ cells. Nat Commun 12(1): 892. PubMed ID: 33563972
Marcillac, F., Bousquet, F., Alabouvette, J., Savarit, F. & Ferveur, J. F. (2005). A mutation with major effects on Drosophila melanogaster sex pheromones. Genetics 171: 1617-1628. PubMed Citation: 15489528
Marin, I., Franke, A., Bashaw, G. J. and Baker, B. S. (1996). The dosage compensation system of Drosophila is co-opted by newly evolved X chromosomes. Nature 383: 160-163. PubMed ID: 8774878
Menon, D. U., Coarfa, C., Xiao, W., Gunaratne, P. H. and Meller, V. H. (2014). siRNAs from an X-linked satellite repeat promote X-chromosome recognition in Drosophila melanogaster. Proc Natl Acad Sci U S A 111(46):16460-5. PubMed ID: 25368194
Mercer, M., Dasgupta, A., Pawłowski, K., Buszczak, M. (2025). Bourbon and Mycbp function with Otu to promote Sxl protein expression in the Drosophila female germline. Proc Natl Acad Sci U S A, 122(15):e2426524122 PubMed ID: 40215271
Neville, M. C., Nojima, T., Ashley, E., Parker, D. J., Walker, J., Southall, T., Van de Sande, B., Marques, A. C., Fischer, B., Brand, A. H., Russell, S., Ritchie, M. G., Aerts, S. and Goodwin, S. F. (2014). Male-specific fruitless isoforms target neurodevelopmental genes to specify a sexually dimorphic nervous system. Curr Biol 24: 229-241. PubMed ID: 24440396
Ota, R., Hayashi, M., Morita, S., Miura, H. and Kobayashi, S. (2021). Absence of X-chromosome dosage compensation in the primordial germ cells of Drosophila embryos. Sci Rep 11(1): 4890. PubMed ID: 33649478
Primus, S., Pozmanter, C., Baxter, K. and Van Doren, M. (2019). Tudor-domain containing protein 5-like promotes male sexual identity in the Drosophila germline and is repressed in females by Sex lethal. PLoS Genet 15(7): e1007617. PubMed ID: 31329582
Ramakrishnan, P., Joshi, A., Tulasi, M. and Yadav, P. (2022). Monochromatic visible lights modulate the timing of pre-adult developmental traits in Drosophila melanogaster. Photochem Photobiol Sci. PubMed ID: 36583814
Ramírez, F.,Lingg, T., Toscano, S., Lam, K.C., Georgiev, P., Chung, H.R., Lajoie,
B.R., de Wit, E., Zhan, Y., de Laat, W., Dekker, J., Manke, T. and Akhtar, A. (2015). High-affinity sites form an interaction network to facilitate spreading of the MSL complex across the X chromosome in Drosophila. Mol Cell 60: 146-162. PubMed ID: 26431028
Rideout, E. J., Narsaiya, M. S. and Grewal, S. S. (2015). The sex determination gene transformer regulates male-female differences in Drosophila body size. PLoS Genet 11(12): e1005683. PubMed ID: 26710087
Rieder, L. E., Jordan, W. T., 3rd, Larschan, E. N. (2019). Targeting of the Dosage-Compensated Male X-Chromosome during Early Drosophila Development. Cell Rep, 29(13):4268-4275 e4262 PubMed ID: 31875538
Saccone, G., Peluso, I., Artiaco, D., Giordano, E., Bopp, D., et
al. (1998). The Ceratitis capitata homologue of the Drosophila sex-determining gene sex-lethal is structurally conserved, but not sex-specifically regulated. Development 125: 1495-1500. Medline abstract: 9502730
Sato, K., Ito, H. and Yamamoto, D. (2020). teiresias, a Fruitless target gene encoding an immunoglobulin-superfamily transmembrane protein, is required for neuronal feminization in Drosophila. Commun Biol 3(1): 598. PubMed ID: 33087851
Sawala, A. and Gould, A. P. (2017). The sex of specific neurons controls female body
growth in Drosophila. PLoS Biol 15(10): e2002252. PubMed ID: 28976974
Schauer, T., Ghavi-Helm, Y., Sexton, T., Albig, C., Regnard, C., Cavalli, G., Furlong, E. E. and Becker, P. B. (2017). Chromosome topology guides the Drosophila Dosage Compensation Complex for target gene activation. EMBO Rep. PubMed ID: 28794204
Soruco, M. M., Chery, J., Bishop, E. P., Siggers, T., Tolstorukov, M. Y., Leydon, A. R., Sugden, A. U., Goebel, K., Feng, J., Xia, P., Vedenko, A., Bulyk, M. L., Park, P. J. and Larschan, E. (2013). The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes Dev 27(14): 1551-1556. PubMed ID: 23873939
Sturgill, D., Zhang, Y., Parisi, M. and Oliver, B. (2007). Demasculinization of X chromosomes in the Drosophila genus. Nature 450: 238-241. PubMed Citation: 17994090
Sun, X., Yang, H., Sturgill, D., Oliver, B., Rabinow, L. and Samson, M. L. (2015). Sxl-dependent, tra/tra2-independent alternative splicing of the Drosophila melanogaster X-Linked gene found in neurons. G3 (Bethesda) 5(12):2865-74. PubMed ID: 26511498
Testa, N. D., Ghosh, S. M. and Shingleton, A. W. (2013). Sex-specific weight loss mediates sexual size dimorphism in Drosophila melanogaster. PLoS One 8(3): e58936. PubMed ID: 23555608
Tikhonova, E., Mariasina, S., Efimov, S., Polshakov, V., Maksimenko, O., Georgiev, P. and Bonchuk, A. (2022). Structural basis for interaction between CLAMP and MSL2 proteins involved in the specific recruitment of the dosage compensation complex in Drosophila. Nucleic Acids Res 50(11): 6521-6531. PubMed ID: 35648444
Urban, J. A., Doherty, C. A., Jordan, W. T., Bliss, J. E., Feng, J., Soruco, M. M., Rieder, L. E., Tsiarli, M. A. and Larschan, E. N. (2016). The essential Drosophila CLAMP protein differentially regulates non-coding roX RNAs in male and females. Chromosome Res [Epub ahead of print]. PubMed ID: 27995349
Valsecchi, C. I. K., Marois, E., Basilicata, M. F., Georgiev, P. and Akhtar, A. (2021). Distinct mechanisms mediate X chromosome dosage compensation in Anopheles and Drosophila. Life Sci Alliance 4(9). PubMed ID: 34266874
Vedelek, V., Kovacs, A. L., Juhasz, G., Alzyoud, E. and Sinka, R. (2021). The tumor suppressor archipelago E3 ligase is required for spermatid differentiation in Drosophila testis. Sci Rep 11(1): 8422. PubMed ID: 33875704
Villa, R., Schauer, T., Smialowski, P., Straub, T. and Becker, P. B. (2016). PionX sites mark the X chromosome for dosage compensation. Nature 537: 244-248. PubMed ID: 27580037
Villa, R., Jagtap, P. K. A., Thomae, A. W., Campos Sparr, A., Forne, I., Hennig, J., Straub, T. and Becker, P. B. (2021). Divergent evolution toward sex chromosome-specific gene regulation in Drosophila. Genes Dev 35(13-14): 1055-1070. PubMed ID: 34140353
Watanabe, T. (2019). Evolution of the neural sex-determination system in insects: does fruitless homolog regulate neural sexual dimorphism in basal insects?. Insect Mol Biol. PubMed ID: 31066110
Wawersik, M., Milutinovich, A., Casper, A. L., Matunis, E., Williams, B. and Van Doren, M. (2005). Somatic control of germline sexual development is mediated by the JAK/STAT pathway. Nature 436: 563-567. PubMed Citation: 16049490
Wehr Mathews, K., Cavegn, M. and Zwicky, M. (2017). Sexual dimorphism of body size is controlled by dosage of the X-chromosomal gene Myc and by the sex-determining gene tra in Drosophila. Genetics 205(3):1215-1228. PubMed ID: 28064166
Wei, K. H., Chatla, K., Bachtrog, D. (2024). Single-cell RNA-seq of Drosophila miranda testis reveals the evolution and trajectory of germline sex chromosome regulation. PLoS Biol, 22(4):e3002605 PubMed ID: 38687805
Witt, E., Shao, Z., Hu, C., Krause, H. M. and Zhao, L. (2021). Single-cell RNA-sequencing reveals pre-meiotic X-chromosome dosage compensation in Drosophila testis. PLoS Genet 17(8): e1009728. PubMed ID: 34403408
Yan, D. and Perrimon, N. (2015). spenito is required for sex determination in Drosophila melanogaster. Proc Natl Acad Sci 112(37): 11606-11. PubMed ID: 26324914
Yang, S. Y., Baxter, E. M. and Van Doren, M. (2012). Phf7 controls male sex determination in the Drosophila germline. Dev Cell 22(5): 1041-1051. PubMed ID: 22595675
Yang, S. Y. (2021). Germline masculinization by Phf7 in D. melanogaster requires its evolutionarily novel C-terminus and the HP1-family protein HP1D3csd. Sci Rep 11(1): 6308. PubMed ID: 33737548
Yatsu, J., Hayashi, M., Mukai, M., Arita, K., Shigenobu, S. and Kobayashi, S. (2008). Maternal RNAs encoding transcription factors for germline-specific gene expression in Drosophila embryos. Int. J. Dev. Biol. 52: 913-923. PubMed Citation: 18956321
Zhou, Q., Ellison, C. E., Kaiser, V. B., Alekseyenko, A. A., Gorchakov, A. A. and Bachtrog, D. (2013). The epigenome of evolving Drosophila neo-sex chromosomes: dosage compensation and heterochromatin formation. PLoS Biol 11: e1001711. PubMed ID: 24265597
Zhou, H., Whitworth, C., Pozmanter, C., Neville, M. C. and Van Doren, M. (2021). Doublesex regulates fruitless expression to promote sexual dimorphism of the gonad stem cell niche. PLoS Genet 17(3): e1009468. PubMed ID: 33788836
Home page: The
Interactive Fly © 2017 Thomas B. Brody, Ph.D.
The Interactive Fly resides on the
Society for Developmental Biology's Web server.