InteractiveFly: GeneBrief
Exchange factor for Arf 6: Biological Overview | References
|
Gene name - Exchange factor for Arf 6
Synonyms - Cytological map position - 94B5-94B6 Function - signaling Keywords - directly inhibits microtubule growth - a cortical collapse factor acting at the plasma membrane - regulates axon growth |
Symbol - Efa6
FlyBase ID: FBgn0051158 Genetic map position - chr3R:22,587,755-22,608,471 NCBI classification - Sec7 and PH_EFA6 domain-containing protein Cellular location - cytoplasmic |
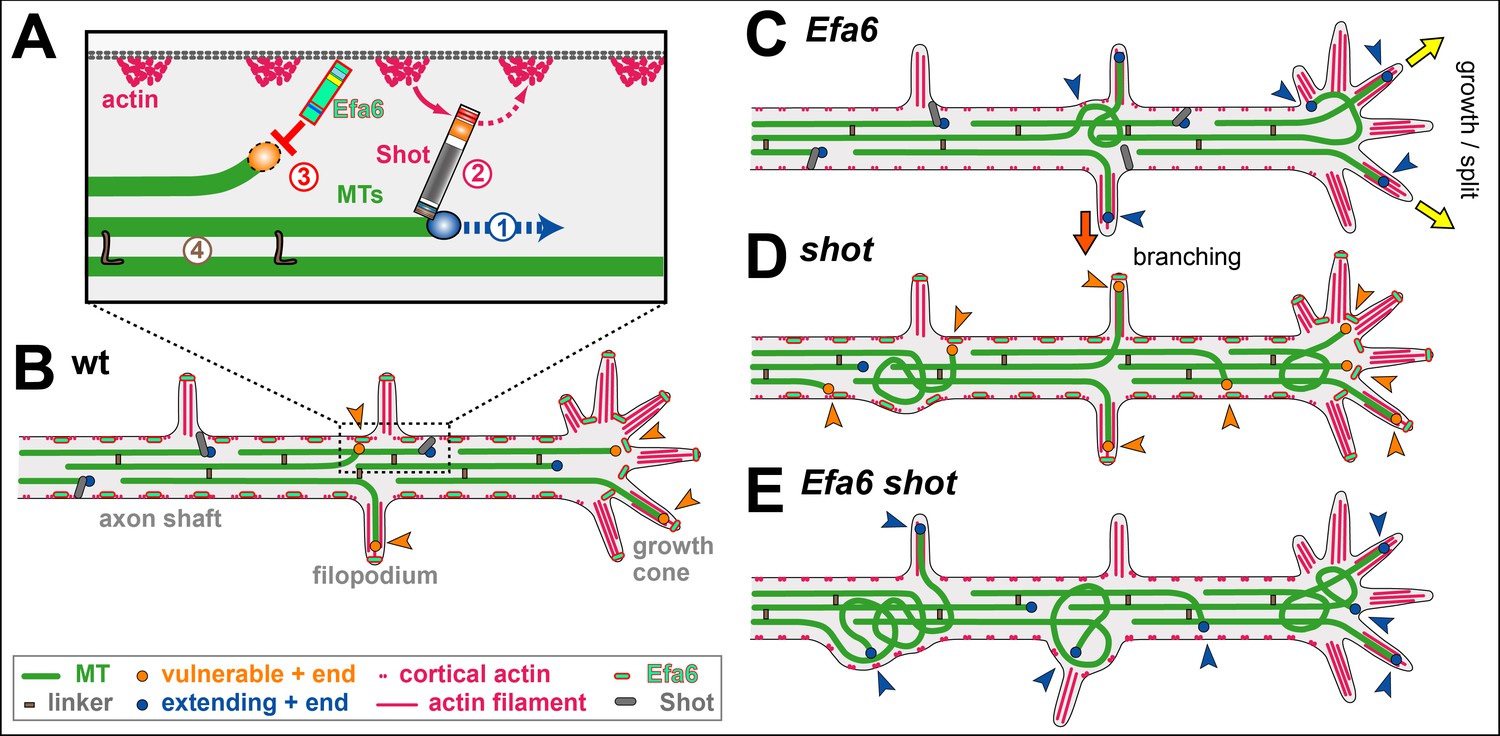
Cortical collapse factors affect microtubule (MT) dynamics at the plasma membrane. They play important roles in neurons, as suggested by inhibition of axon growth and regeneration through the Arf activator Efa6 in C. elegans, and by neurodevelopmental disorders linked to the mammalian kinesin Kif21A. How cortical collapse factors influence axon growth is little understood. This study focussed on the function of Drosophila Efa6 in experimentally and genetically amenable fly neurons. First, it was shown that Drosophila Efa6 can inhibit MTs directly without interacting molecules via an N-terminal 18 amino acid motif (MT elimination domain/MTED) that binds tubulin and inhibits microtubule growth in vitro and cells. If N-terminal MTED-containing fragments are in the cytoplasm they abolish entire microtubule networks of mouse fibroblasts and whole axons of fly neurons. Full-length Efa6 is membrane-attached, hence primarily blocks MTs in the periphery of fibroblasts, and explorative MTs that have left axonal bundles in neurons. Accordingly, loss of Efa6 causes an increase of explorative MTs: in growth cones they enhance axon growth, in axon shafts they cause excessive branching, as well as atrophy through perturbations of MT bundles. Efa6 over-expression causes the opposite phenotypes. Taken together, this work conceptually links molecular and sub-cellular functions of cortical collapse factors to axon growth regulation and reveals new roles in axon branching and in the prevention of axonal atrophy. Furthermore, the MTED delivers a promising tool that can be used to inhibit MTs in a compartmentalised fashion when fusing it to specifically localising protein domains (Qu, 2019).
Axons are the cable-like neuronal extensions that wire the nervous system. They are only 0.1-15 μm in diameter, but can be up to a meter long in humans. It is a fascinating challenge to understand how axons can extend over these enormous distances and branch in orderly manners, but also how these delicate structures can be maintained for a lifetime, that is many decades in humans. It is not surprising that humans gradually lose about 40% of their axons towards old age, and that axon decay is a prominent neurodegenerative phenomenon (Qu, 2019).
Essential for axon biology are the parallel bundles of microtubules (MTs) running all along the axon shaft; these bundles provide (1) structural support, (2) highways for life-sustaining cargo transport, and (3) a source of MTs that can leave these bundles to drive morphogenetic changes. Through being organised in this way, MTs essentially drive processes of axon growth, branching and maintenance. The dynamics of MTs are orchestrated through MT-binding and -regulating proteins, for most of what is known about the molecular mechanisms of function. However, such knowledge alone is usually not sufficient to explain their cellular roles (Qu, 2019).
For example, cortical collapse factors are cell surface-associated proteins which specifically inhibit MTs that approach the cell periphery. Previous reports suggested important roles for cortical collapse factors in regulating axon growth: the ARF activator Efa6 (exchange factor for ARF6) in C. elegans negatively impacts on developmental and regenerative axon growth (Chen, 2015; Chen, 2011; O'Rourk, 2010); the mammalian type four kinesin KIF21A also affects axon growth and links to the neurodevelopmental eye movement disorder 'congenital fibrosis of extraocular muscles' (OMIM reference #135700). However, it can currently only be hypothesized how the molecular functions of these two collapse factors link to axon growth, most likely by acting in growth cones (GCs) (Qu, 2019).
GCs are the amoeboid tip structures through which axons extend to wire the nervous system during development or regeneration. The axonal MT bundles terminate in the centre of GCs; from here, single MTs splay into the actin-rich periphery of GCs. These explorative MTs can trigger extension of the entire MT bundle into their direction, thus elongating the axon; by inhibiting such explorative MTs, cortical collapse factors could negatively impact on axon growth (Qu, 2019).
In line with this argument, and depending on where cortical collapse factors are present and functionally active, further functional predictions could be made: for example, collateral branching of axons along their shafts has been described to depend on explorative MTs that leave the parallel axonal bundles and polymerise towards the periphery. Cortical collapse factors might therefore be negative regulators of axon branching (Qu, 2019).
Other roles might concern axon maintenance: the model of 'local axon homeostasis' states that the force-enriched environment in axons biases MTs to buckle or project out of the bundle to seed pathological areas of MT disorganisation. By inhibiting off-track MTs in the axon shaft, cortical collapse factors might prevent such processes, acting in parallel to other bundle-maintaining factors. For example, spectraplakins serve as spacers that keep polymerising MTs away from the cortex by linking the tips of extending MTs to the axonal surface and guiding them into parallel bundles. Their deficiency in any organism causes severe MT disorganisation, potentially explaining human dystonin-linked HSAN6 ('type six hereditary sensory and autonomic neuropathy'; OMIM ID: 614653). If the hypothesis is correct, loss of cortical collapse factors in axon shafts would also cause MT disorganisation, but through a very different mechanistic route (Qu, 2019).
This study makes use of Drosophila neurons as a well-established, powerful model for studying roles of MT regulators. Using in vitro and cellular assays, this study showed that Drosophila Efa6 is a cortical collapse factor acting through its N-terminal MT elimination domain (MTED). The MTED binds tubulin and blocks MT polymerisation in vitro which indicates that the effect of the peptide is due to a direct interaction between the peptide and tubulin. By localising to neuronal membranes, it only abolishes explorative MTs. This subcellular role translates into negative regulation of axon growth and branching and the prevention of pathological MT disorganisation, both in cultured neurons and in vivo. It is proposed that Efa6 functions as a quality control or axonal maintenance factor that keeps explorative MTs in check, thus playing a complementary role to spectraplakins that prevent MTs from leaving axonal bundles (Qu, 2019).
Axons are the structures that wire the brain and body and are therefore fundamental to nervous system function. To understand how axons are formed during development, can be maintained in a plastic state thereafter, and why they deteriorate in pathological conditions, it is necessary to increase knowledge of axonal cell biology. The MT bundles that form the core of axons are an essential aspect of this cell biology, and understanding how these bundles are regulated and contribute to axon morphogenesis will provide essential insights into axon development and maintenance. This study has addressed fundamental contributions made by cortical collapse factors. This work started from reports that two such factors from distinct protein families both negatively impact on axon growth in species as diverse as C. elegans (CeEfa6) and mouse (Kif21A) (Qu, 2019).
This study found that DmEfa6 likewise acts as a negative regulator of axon growth. Efa6 is a cortical collapse factor, inhibiting MTs primarily via the 18aa long MTED. Since the MTED is the only shared motif with CeEfa6 in an otherwise entirely divergent N-terminus, this clearly demonstrates that the MTED is functionally conserved between both species (Qu, 2019).
Capitalising on Drosophila neurons as a conceptually well-established model for studies of axonal MT regulation, two novel roles were demonstrated for Efa6: as a negative regulator of axon branching and a quality control factor maintaining MT bundle organisation. To perform these functions, Efa6 does not affect the dynamics of MTs contained within the central axonal bundles, but it inhibits mainly those MTs that leave these bundles (see A model for axonal roles of Efa6). By inhibiting explorative MTs in GCs, it negatively impacts on a key event underlying axon growth. By inhibiting off-track MTs in the axon shaft, it tones down the machinery that seeds new interstitial branches, but also prevents these MTs from going astray and causing MT disorganisation (Qu, 2019).
Therefore, this work provides conceptual understanding of cortical collapse factors, which can explain how their molecular functions and subcellular roles in MT regulation link to their reported axonal growth phenotypes during development and regeneration, and to their additional functions in axon branching and maintenance. Apart from existing links of cortical collapse factors to neurodevelopmental disorders, it is therefore predicted that future links will be made to neurodegeneration (Qu, 2019).
During axon growth, MTs constantly polymerise towards the periphery of GCs; the advance of many of these MTs is inhibited at the leading edge, and this work shows that cortical collapse factors are key mediators to this end. Only a fraction of MTs enters filopodia, potentially helped by active guidance mechanisms such as MT-actin cross-linkage (e.g. through spectraplakins, tau, drebrin-EB3). The widely accepted protrusion-engorgement-consolidation model of axon growth proposes that stabilised MTs in filopodia can seed axon elongation events. This model is consistent with the current findings for Efa6. Thus loss of Efa6 can contribute to enhanced axon growth in two ways: firstly, through allowing more MTs to enter filopodia; secondly, by allowing them to dwell in filopodia for longer, thus enhancing the likelihood of their stabilisation. This scenario can explain why loss of Efa6 in C. elegans improves axon re-growth after injury and growth overshoot during development, and why the higher levels of Kif21A levels in GCs causes stalled axon growth (Qu, 2019).
In C. elegans it was shown that axonal injury leads to a re-localisation of CeEfa6 to MT minus ends in the axon core (Chen, 2015). None of the conditions used in the current study reproduced such behaviour with fly Efa6. Furthermore, it was shown that such central pools of CeEfa6 require their MTED to recruit two kinases: TAC-1 (homologue of TACC/transforming-acidic-coiled-coil) and ZYG-8 (homologue of DCLK/Doublecortin-Like-Kinase; Chen, 2015). However, in contrast to Efa6, both of these kinases perform growth-enhancing functions and play a secondary, delayed role downstream of Efa6. They are therefore unsuited to explain the direct MT-inhibiting roles of the MTED. In contrast, virtually all structure-function analyses performed with CeEfa6 in developing and regenerating axons perfectly match the data and can be explained through the proposed model. Based on these findings, one might argue that CeEfa6 detachment from the membrane could be the consequence of injury-induced physiological changes that would then pose a threat to axonal MT bundles; localisation to MT minus ends could therefore represent a protective sequestration mechanism. Another C. elegans study reported that loss of Efa6 has no impact on MT length in developing axons (Yogev, 2016), which appears consistent with the current data. They also found an increase in MT numbers, but there is currently no mechanism to explain this in non-injury conditions where CeEfa6 stays at the membrane (Chen, 2015; Qu, 2019 and references therein).
Interestingly, mammalian Efa6 also plays a role in axon regeneration. However, this mechanism is entirely different, in that it requires the C-terminus to activate Arf6 which, in turn, regulates integrin trafficking at the axon initial segment (Eva, 2017; Qu, 2019 and references therein).
Axon branching can occur via GC split, in that diverging MTs get stabilised in parallel in the same GC. Alternatively, it can occur through interstitial branching which involves the active generation (e.g. through MT severing) and then stabilisation of off-track MTs. Both models agree with observations in Efa6-deficient/over-expressing neurons: greater/lower numbers of MTs were found in GC and shaft filopodia at 6 hours in vitro, which then correlate with enhanced/reduced axonal branch numbers in mature neurons (Qu, 2019).
If interstitial branch formation is negatively regulated by Efa6, this poses the question as to whether Efa6 has to be actively down-regulated in healthy neurons for branching to occur. Efa6 could either be physically removed from future branch points or its MT inhibition function could be switched off. However, no such regulation appears to be required because Efa6 seems to be in a well-balanced equilibrium. Enough Efa6 appears to be present to inhibit occasional, likely accidental off-track MTs; this capacity is surpassed when the number of off-track MTs is actively increased, for example through MT severing proteins during axonal branch formation. Such a saturation model is supported by experiments with shot: filopodial MT numbers are elevated in shot mutant neurons, although Efa6 is present and functional (as demonstrated by the further increase in filopodial MT numbers in shot Efa6 double-mutant neurons). This is consistent with a model where Efa6 function occurs at a level that is easily saturated when increasing the number of explorative MTs. Such a view would also explain why loss of CeEfa6 promotes axon regeneration in C. elegans, in that the constant base-line of MT inhibition present in the wild-type, is removed in the mutant condition, thus favouring growth-mediating explorative MTs (Qu, 2019).
Axonal MT disorganisation in Efa6-deficient neurons occurs gradually and can even be induced by knock-down of Efa6 at mature stages. Therefore, Efa6 appears to prevent MT disorganisation during axon development and maintenance, as is consistent with its continued expression in the nervous system. Such a continued role makes sense in a scenario where MT bundles remain highly dynamic throughout a neuron's lifetime, constantly undergoing polymerisation to drive renewal processes that prevent senescence (Qu, 2019).
Based on these findings, it is proposed that Efa6 acts as a quality control or maintenance factor within a model of 'local axon homeostasis' (Hahn, 2019; Prokop, 2016). This model states that MTs in the force-enriched environment of axons have a tendency to go off-track and curl up, thus potentially seeding MT disorganisation. Different classes of MT-binding regulators, amongst them spectraplakins, prevent this by actively promoting the bundled conformation. It is proposed that cortical collapse factors act in a complementary way to spectraplakins in that they play no role in maintaining MTs in bundles, but they inhibit those MTs that have escaped the bundling mechanisms (Qu, 2019).
In this scenario, MTs are protected from cortical collapse as long as they are actively maintained in axonal bundles; this can explain the long known conundrum of how axonal MTs extend hundreds of micrometres in relative proximity to the cell cortex in axons, whereas in non-neuronal cells cortical proximity of MTs tends to trigger either their inhibition or tethered stabilisation (Qu, 2019).
This study found that the MTED motif correlates well with MT inhibiting functions of Efa6 family members, whereas the rest of the N-terminus bears no obvious further similarity. Experiments with N-terminal protein and synthetic MTED peptide, both reveal association with MTs/tubulin. The MTED strongly interferes with MT polymerisation. Future co-crystallisation experiments are required to reveal how the MTED works. Given its small size it is hypothesised that it simply blocks assembly, rather than acting via more complex mechanisms such as active promotion of depolymerisation (e.g., kinesin-8 and -13, XMap215) or severing (e.g., spastin, katanin, fidgetin (Qu, 2019).
In any case, the small size of MTEDs might come in handy as experimental tools to inhibit MTs, potentially displaying complementary properties to existing genetic tools such as the kinesin-13 Kif2C, stathmin or spastin. Importantly, the experiments with the CAAX domain have shown that Efa6's MT inhibiting function can be targeted to specific subcellular compartments to clear them of MTs, thus opening up a wide range of future applications (Qu, 2019).
Interestingly, the MT-inhibiting role of Efa6 seems not to be conserved in chordates when taking the MTED as indicator for this function. However, roles of cortical collapse factors in neurons seem to have been taken over by other proteins such as the kinesin-4 family member Kif21A. The CFEOM1-linked Kif21AR954W mutation causes the protein to relocate from the axon shaft to the growth cone of cultured hippocampal neurons. In consequence, increased Kif21A levels in GCs cause reduced axon growth - and this study observed the same with Efa6 over-expression. The decreased levels of Kif21A in proximal axons correlate with a local increase in side branches - and the same is observed with Efa6 loss of function (Qu, 2019).
Finally, this study found that the C-terminal domains of Efa6 might display some degree of functional conservation. So far, work on mammalian PSDs has revealed functions for C-terminal domains in regulating ARF6, ARF1 or ARL14 during actin cytoskeletal reorganisation and membrane ruffling, tumour formation, axon regeneration and immune regulation. The finding that PSD1 and C-terminal Efa6 constructs cause similar membrane ruffling phenotypes in fibroblasts, suggests that some conserved functions reside in this region and might further contribute, together with N-terminally mediated MT inhibition, to the neuronal or non-neuronal defects that cause semi-lethality displayed by Efa6 mutant flies (Qu, 2019).
It is proposed that Efa6 acts as a cortical collapse factor which is important for the regulation of axonal MTs and relevant for axon growth, maintenance and branching. Although this function of Efa6 is evolutionarily not widely conserved, these findings provide a helpful paradigm for studies of other classes of cortical collapse factors also in mammalian neurons. Promising research avenues will be to refine the mechanistic understanding of how Efa6 blocks MT polymerisation, not only to better understand how it can be regulated in axons, but also to better exploit MTEDs as molecular tools in cell biological research (Qu, 2019).
It has long been thought that microtubule disassembly, one of the earliest cellular events, contributes to neuronal pruning and neurodegeneration in development and disease. However, how microtubule disassembly drives neuronal pruning remains poorly understood. This study conducted a systematic investigation of various microtubule-destabilizing factors and identified exchange factor for Arf6 (Efa6) and Stathmin (Stai) as new regulators of dendrite pruning in ddaC sensory neurons during Drosophila metamorphosis. Efa6 is both necessary and sufficient to regulate dendrite pruning. Interestingly, Efa6 and Stai facilitate microtubule turnover and disassembly prior to dendrite pruning without compromising the minus-end-out microtubule orientation in dendrites. Moreover, pharmacological and genetic manipulations strongly support a key role of microtubule disassembly in promoting dendrite pruning. Thus, this systematic study highlights the importance of two selective microtubule destabilizers in dendrite pruning and substantiates a causal link between microtubule disassembly and neuronal pruning (Bu, 2021).
Microtubule disassembly, one of the earliest cellular alterations in pruning dendrites or axons, has been thought to be a prerequisite for the execution of neuronal pruning. This systematic study reveals that dendrite pruning selectively requires two microtubule destabilizers, Efa6 and Stai. Moreover, pharmacological and genetic manipulations strongly support a causative role of microtubule disassembly in promoting dendrite pruning of ddaC sensory neurons (Bu, 2021).
The Efa6 protein family is conserved from yeast to mammals and was originally identified in mammals to regulate endosomal membrane recycling and actin cytoskeletal rearrangement via its C-terminal Sec7 GEF domain. In the mammalian nervous systems, Efa6, via the GEF domain, regulates dendritic spine formation and axon regeneration in an Arf6-dependent manner. However, in worms and flies, Efa6 orthologs act independently of Arf6 to inhibit microtubule polymerization via their respective N-terminal MTED domains. The MTED domain is conserved in worms and flies, but not in mammals. Through their MTEDs, Efa6s function as negative regulators of developmental axon growth/branching and axonal regeneration after injury. This study reports an important role of Efa6 in promoting dendrite pruning of sensory neurons via its MTED domain. Multiple genetic manipulations with loss or gain of Efa6 function demonstrate that Efa6 is both necessary and sufficient to promote dendrite pruning. Moreover, the structure-function analysis reveals that the MTED domain is essential for Efa6 to promote dendrite pruning, whereas the C-terminal Sec7 GEF domain is dispensable. Overexpression of the MTED-deleted Efa6 variant was unable to induce precocious dendrite pruning phenotype. Unlike other microtubule regulators, such as Patronin and Msps/TACC, Efa6 does not modulate the minus-end-out microtubule orientation in ddaC dendrites, but rather inhibits microtubule growth and promotes microtubule disassembly at the proximal regions of the dendrites (Bu, 2021).
How does Efa6 disassemble microtubules in the dendrites during pruning? This study found that loss of Efa6 function resulted in increased number of EB1-GFP-labelled plus ends of microtubules and reduced microtubule depolymerization/turnover at the proximal dendrites, whereas gain of Efa6 function led to enhanced microtubule depolymerization and disassembly. The model that Efa6 may directly disassemble microtubules in vivo at the proximal dendrites is preferred, leading to initial severing of dendrites. In line with this model, fly Efa6 directly interacts with tubulins and inhibits microtubule polymerization per se. In the case of axon injury and regeneration in nematodes, Efa6 was reported to indirectly inhibit microtubule dynamics by suppressing its binding partners, TAC-1 (TACC in Drosophila) and ZYG-8 (Doublecortin-like kinase, CG17528 in Drosophila;). However, fly TACC and Efa6 may function independently during dendrite pruning. A previous study reported that TACC regulates the minus-end-out microtubule orientation to promote dendrite pruning in ddaC dendrites. By contrast, Efa6 does not affect dendritic microtubule orientation in these neurons. Moreover, mutants of the fly zyg8/CG17528 were generated that did not show any dendrite pruning defect, and double mutant of efa6 and zyg8/CG17528 displayed no genetic interaction. Efa6 has a cortical localization pattern in worm neurons/embryos. Interestingly, the cytoplasmic distribution of fly Efa6 in ddaC neurons was found despite its membrane association in epidermal cells. This finding raises the possibility that Efa6 might be released from the plasma membrane to the cytoplasm where it might more potently eliminate microtubules during dendrite pruning. In support of this notion, worm Efa6 can relocalize from the plasma membrane to microtubule minus ends and inhibit microtubule dynamics in axons upon injury (Bu, 2021).
This study has systematically interrogated various microtubule-destabilizing factors for their possible roles in dendrite pruning using both RNAi and clonal approaches. Dendrite pruning was demonstrated to require two microtubule destabilizers, Efa6 and Stai. Stai and its mammalian homologues negatively regulate microtubule dynamics and disassemble microtubules via a mechanism distinct from Efa6. Stai can either sequester α/β-tubulin dimers and prevent their incorporation into growing microtubules or directly interact with microtubules to promote their disassembly. Stai has been involved in diverse models of neurodegeneration, neuronal migration axonal transport, neuronal polarization and regeneration as well as plasticity. Drosophila Stai regulates axonal microtubule integrity, synapse stability and neuronal functions. This study identified an additive function of Stai and Efa6 in dendrite pruning of ddaC neurons. Like Efa6, Stai promotes microtubule turnover/disassembly but does not regulate microtubule orientation in the dendrites of ddaC sensory neurons. Loss of Stai function resulted in an increase in polymerized microtubules; conversely, overexpression of Stai caused strong reductions in microtubule growth and mass. Thus, Stai is essential for inhibiting microtubule polymerization and promoting the disassembly during dendrite pruning. However, in contrast to this finding, a previous study has also reported that loss of Stai function results in decreased microtubule levels in the axons of Drosophila CNS neurons. It is possible that differential roles of Stai in PNS and CNS neurons might be due to its different phosphorylation. In Xenopus, the microtubule-destabilizing activity of Stai is dependent on its phosphorylation state, which is regulated by the serine/threonine type-2A phosphatase (Bu, 2021).
The microtubule-severing enzymes (katanin, fidgetin and spastin), which hydrolyse ATP to sever microtubules into small pieces in vitro, are apparent candidates that disassemble dendritic microtubules to promote neuronal pruning. These enzymes were also reported to sever microtubules and regulate the neuromuscular junction development or dendrite arborization in Drosophila and mammals . Moreover, spastin was reported to promote axon pruning at the neuromuscular junctions in postnatal mice by locally destabilizing microtubules. However, RNAi knockdown of these severing enzymes did not cause any prominent dendrite pruning defects in Drosophila ddaC neurons in the previous studies. To exclude the possibility that RNAi knockdown was inefficient for these genes, several mutant alleles were generated for these genes, and their clonal analysis was conducted in this study. The results further substantiate that these individual enzymes are indeed dispensable for dendrite pruning. Kat-60L1, an AAA ATPase analogous to Kat-60, is involved in ddaC dendrite pruning. However, Kat-60L1 had no apparent microtubule-disassembly function in ddaC neurons. Unlike Kat-60, overexpression of the Kat-60L1 short isoform neither impaired microtubule turnover nor affected the microtubule levels in the dendrites of ddaC neuron. The effect of the long isoform of Kat-60L1 in ddaC neurons was also analyzed, as the long isoform, but not the short isoform, was reported to possess microtubule-disassembly function in adult mechanosensory neurons. Unexpectedly, similar to the short isoform, reduced Futsch-positive microtubules were not observed in the dendrites when the long isoform of Kat-60L1 was overexpressed in ddaC neurons. Moreover, no significant alteration in the levels of polymerized microtubules was observed in dendrites and soma of kat-60L1F1 mutant ddaC neurons. Thus, none of the data suggest that Kat-60L1 acts as a microtubule-severing enzyme in ddaC neurons, which contrasts with a recent finding showing that purified Kat-60L1 protein possesses the microtubule-severing activity in vitro. However, the data cannot rule out the possibility that Kat-60L1 promotes dendrite pruning via severing dendritic microtubules. Further studies will be necessary to determine its potential microtubule-severing function during ddaC dendrite pruning (Bu, 2021).
Microtubule disassembly precedes membrane scission during neuronal pruning. This study has solated two microtubule-destabilizing factors, Efa6 and Stai, whose mutants exhibited excessive microtubule polymerization without affecting microtubule orientation in dendrites. These findings further provided the opportunity to investigate a relationship between microtubule disassembly and dendrite pruning. First, efa6 RNAi larvae were treated with colchicine, a microtubule-destabilizing drug. A low concentration of colchicine not only reduced microtubule polymerization but also fully rescued the dendrite pruning defects in efa6 loss-of-function neurons. Second, the treatment with the microtubule-stabilizing drug taxol caused excessive microtubule polymerization in dendrites and inhibited dendrite pruning in wild-type neurons. Third, taxol treatment significantly enhanced dendrite pruning defects in the efa6 RNAi background. Finally, reduced γ-tubulin in efa6 mutant neurons, which likely downregulates microtubule nucleation and in turn polymerization, almost fully rescued the pruning defects. Taken together, multiple lines of genetic and pharmacological evidence support that microtubule disassembly drives dendrite pruning in ddaC sensory neurons (Bu, 2021).
In summary, this systematic study highlights important roles of two negative regulators of microtubule polymerization, Efa6 and Stai, in facilitating dendrite pruning via microtubule disassembly. Moreover, this study supports a causal relationship between microtubule disassembly and dendrite pruning during early metamorphosis (Bu, 2021).
Ubiquitously expressed genes have been implicated in a variety of specific behaviors, including responses to ethanol. However, the mechanisms that confer this behavioral specificity have remained elusive. Previously, it has been shown that the ubiquitously expressed small GTPase Arf6 is required for normal ethanol-induced sedation in adult Drosophila. This study shows that this behavioral response also requires Efa6, one of (at least) three Drosophila Arf6 guanine exchange factors. Ethanol-naive Arf6 and Efa6 mutants were sensitive to ethanol-induced sedation and lacked rapid tolerance upon re-exposure to ethanol, when compared with wild-type flies. In contrast to wild-type flies, both Arf6 and Efa6 mutants preferred alcohol-containing food without prior ethanol experience. An analysis of the human ortholog of Arf6 and orthologs of Efa6 (PSD1-4) revealed that the minor G allele of single nucleotide polymorphism (SNP) rs13265422 in PSD3, as well as a haplotype containing rs13265422, was associated with an increased frequency of drinking and binge drinking episodes in adolescents. The same haplotype was also associated with increased alcohol dependence in an independent European cohort. Unlike the ubiquitously expressed human Arf6 GTPase, PSD3 localization is restricted to the brain, particularly the prefrontal cortex (PFC). Functional magnetic resonance imaging revealed that the same PSD3 haplotype was also associated with a differential functional magnetic resonance imaging signal in the PFC during a Go/No-Go task, which engages PFC-mediated executive control. This translational analysis, therefore, suggests that PSD3 confers regional specificity to ubiquitous Arf6 in the PFC to modulate human alcohol-drinking behaviors (Gonzalez, 2018).
Membrane trafficking establishes and maintains epithelial polarity. Rab22a has a polarized distribution in activated T-cells, but its role in epithelial polarity has not been investigated. Previous work showed that Rab14 acts upstream of Arf6 to establish the apical membrane initiation site (AMIS), but its interaction with Rab22a is unknown. This study shows that Rab14 and Rab22a colocalize in endosomes of both unpolarized and polarized MDCK cells and Rab22a localizes to the cell:cell interface of polarizing cell pairs. Knockdown of Rab22a results in a multi-lumen phenotype in three-dimensional culture. Further, overexpression of Rab22a in Rab14 knockdown cells rescues the multi-lumen phenotype observed with Rab14 knockdown, suggesting that Rab22a is downstream of Rab14. Because of the relationship between Rab14 and Arf6, the effect of Rab22a knockdown on Arf6 was investigated. This study found that Rab22a knockdown results in decreased active Arf6 and that Rab22a co-immunoprecipitates with the Arf6 GEF EFA6. In addition, EFA6 is retained in intracellular puncta in Rab22a KD cells. These results suggest that Rab22a acts downstream of Rab14 to traffic EFA6 to the AMIS to regulate Arf6 in the establishment of polarity (Blum, 2020).
The Arf6-specific exchange factor EFA6 is involved in the endocytic/recycling pathway for different cargos. In addition EFA6 acts as a powerful actin cytoskeleton organizer, a function required for its role in the establishment of the epithelial cell polarity and in neuronal morphogenesis. Previous work showed that the C-terminus of EFA6 (EFA6-Ct) is the main domain which contributes to actin reorganization. This study sought to decipher, at the molecular level, how EFA6 controls the dynamic and structuring of actin filaments. EFA6-Ct was shown to interferes with actin polymerization by interacting with and capping actin filament barbed ends. Further, in the presence of actin mono-filaments, the addition of EFA6-Ct triggered the formation of actin bundles. In cells, when the EFA6-Ct was directed to the plasma membrane, as is the case for the full-length protein, its expression induced the formation of membrane protrusions enriched in actin cables. Collectively these data explain, at least in part, how EFA6 plays an essential role in actin organization by interacting with and bundling F-actin (Macia, 2019).
Cell attachment to the extracellular matrix (ECM) requires a balance between integrin internalization and recycling to the surface that is mediated by numerous proteins, emphasizing the complexity of these processes. Upon ligand binding in various cells, the beta1 integrin is internalized, traffics to early endosomes, and is returned to the plasma membrane through recycling endosomes. This trafficking process depends on the cyclical activation and inactivation of small guanosine triphosphatases (GTPases) by their specific guanine exchange factors (GEFs) and their GTPase-activating proteins (GAPs). This study found that the cell surface antigen CD13, a multifunctional transmembrane molecule that regulates cell-cell adhesion and receptor-mediated endocytosis, also promoted cell migration and colocalized with beta1 integrin at sites of cell adhesion and at the leading edge. A lack of CD13 resulted in aberrant trafficking of internalized beta1 integrin to late endosomes and its ultimate degradation. These data indicate that CD13 promoted ARF6 GTPase activity by positioning the ARF6-GEF EFA6 at the cell membrane. In migrating cells, a complex containing phosphorylated CD13, IQGAP1, GTP-bound (active) ARF6, and EFA6 at the leading edge promoted the ARF6 GTPase cycling and cell migration. Together, these findings uncover a role for CD13 in the fundamental cellular processes of receptor recycling, regulation of small GTPase activities, cell-ECM interactions, and cell migration (Ghosh, 2019).
Central nervous system (CNS) axons lose their intrinsic ability to regenerate upon maturity, whereas peripheral nervous system (PNS) axons do not. A key difference between these neuronal types is their ability to transport integrins into axons. Integrins can mediate PNS regeneration, but are excluded from adult CNS axons along with their Rab11 carriers. It was reasoned that exclusion of the contents of Rab11 vesicles including integrins might contribute to the intrinsic inability of CNS neurons to regenerate, and this was investigated by performing laser axotomy. A novel regulator of selective axon transport and regeneration was identified, the ARF6 guanine-nucleotide-exchange factor (GEF) EFA6 (also known as PSD). EFA6 exerts its effects from a location within the axon initial segment (AIS). EFA6 does not localise at the AIS in dorsal root ganglion (DRG) axons, and in these neurons, ARF6 activation is counteracted by an ARF GTPase-activating protein (GAP), which is absent from the CNS, ACAP1. Depleting EFA6 from cortical neurons permits endosomal integrin transport and enhances regeneration, whereas overexpressing EFA6 prevents DRG regeneration. These results demonstrate that ARF6 is an intrinsic regulator of regenerative capacity, implicating EFA6 as a focal molecule linking the AIS, signalling and transport (Eva, 2017).
EFA6D (guanine nucleotide exchange factor for ADP-ribosylation factor 6 [Arf6]D) is also known as EFA6R, Psd3, and HCA67. It is the fourth member of the EFA6 family with guanine nucleotide exchange activity for Arf6, a small guanosine triphosphatase (GTPase) that regulates endosomal trafficking and actin cytoskeleton remodeling. This paper proposes a classification and nomenclature of 10 EFA6D variants deposited in the GenBank database as EFA6D1a, 1b, 1c, 1d, 1s, 2a, 2b, 2c, 2d, and 2s based on the combination of N-terminal and C-terminal insertions. Polymerase chain reaction analysis showed the expression of all EFA6D variants except for variants a and d in the adult mouse brain. Immunoblotting analysis with novel variant-specific antibodies showed the endogenous expression of EFA6D1b, EFA6D1c, and EFA6D1s at the protein level, with the highest expression being EFA6D1s, in the brain. Immunoblotting analysis of forebrain subcellular fractions showed the distinct subcellular distribution of EFA6D1b/c and EFA6D1s. The immunohistochemical analysis revealed distinct but overlapping immunoreactive patterns between EFA6D1b/c and EFA6D1s in the mouse brain. In immunoelectron microscopic analyses of the hippocampal CA3 region, EFA6D1b/c was present predominantly in the mossy fiber axons of dentate granule cells, whereas EFA6D1s was present abundantly in the cell bodies, dendritic shafts, and spines of hippocampal pyramidal cells. These results provide the first anatomical evidence suggesting the functional diversity of EFA6D variants, particularly EFA6D1b/c and EFA6D1s, in neurons (Fukaya, 2016).
Axon injury triggers a series of changes in the axonal cytoskeleton that are prerequisites for effective axon regeneration. In Caenorhabditis elegans the signaling protein Exchange Factor for ARF-6 (EFA-6) is a potent intrinsic inhibitor of axon regrowth. This study shows that axon injury triggers rapid EFA-6-dependent inhibition of axonal microtubule (MT) dynamics, concomitant with relocalization of EFA-6. EFA-6 relocalization and axon regrowth inhibition require a conserved 18-aa motif in its otherwise intrinsically disordered N-terminal domain. The EFA-6 N-terminus binds the MT-associated proteins TAC-1/Transforming-Acidic-Coiled-Coil, and ZYG-8/Doublecortin-Like-Kinase, both of which are required for regenerative growth cone formation, and which act downstream of EFA-6. After injury TAC-1 and EFA-6 transiently relocalize to sites marked by the MT minus end binding protein PTRN-1/Patronin. It is proposed that EFA-6 acts as a bifunctional injury-responsive regulator of axonal MT dynamics, acting at the cell cortex in the steady state and at MT minus ends after injury (Chen, 2015).
ADP-ribosylation factor 6 (ARF6) small GTPase regulates membrane trafficking and cytoskeleton rearrangements at the plasma membrane (PM) by cycling between the GTP-bound active and GDP-bound inactive conformations. Guanine nucleotide exchange factors (GEFs) activate ARF6. The exchange factor for ARF6 (EFA6) R has been identified as a biomarker for ovarian cancer. EFA6R shares the catalytic Sec7, pleckstrin homology (PH), and coiled coil (CC) domains of the other EFA6 family GEFs. This study reports the functional characterization of EFA6R. Endogenous EFA6R was present in the plasma membrane fraction. The exogenously expressed FLAG- and GFP-tagged EFA6R were targeted to the PM. In vitro, GFP-EFA6R associated weakly but preferentially with phosphatidylinositol 4,5-bisphosphate (PIP2) through the PH domain. EFA6R required both its PH and CC domains localized at the C terminus to target the PM. Consistent with this, EFA6R lacking the CC domain (EFA6RDeltaCC) was released from the PM into the cytosol upon PIP2 depletion, whereas EFA6R release from the PM required both PIP2 depletion and actin destabilization. These results suggest that the dual targeting via the PH and CC domains is important for the PM localization of EFA6R. EFA6R specifically catalyzed the GTP loading of ARF6 in mammalian cells. Moreover, EFA6R regulated ARF6 localization and thereby actin stress fiber loss. The GEF activity of EFA6R was dependent on the presence of the Sec7 domain. The PH and CC domains were also required for the in vivo GEF activity of EFA6R but could be functionally replaced by the CAAX motif of K-Ras, suggesting a role for these domains in the membrane targeting of EFA6R (Kanamarlapudi, 2014).
Guanine nucleotide exchange factors (GEFs) of the exchange factor for Arf6 (EFA6), brefeldin A-resistant Arf guanine nucleotide exchange factor (BRAG), and cytohesin subfamilies activate small GTPases of the Arf family in endocytic events. These ArfGEFs carry a pleckstrin homology (PH) domain in tandem with their catalytic Sec7 domain, which is autoinhibitory and supports a positive feedback loop in cytohesins but not in BRAGs, and has an as-yet unknown role in EFA6 regulation. This study analyzed how EFA6A is regulated by its PH and C terminus (Ct) domains by reconstituting its GDP/GTP exchange activity on membranes. This study found that EFA6 has a previously unappreciated high efficiency toward Arf1 on membranes and that, similar to BRAGs, its PH domain is not autoinhibitory and strongly potentiates nucleotide exchange on anionic liposomes. However, in striking contrast to both cytohesins and BRAGs, EFA6 is regulated by a negative feedback loop, which is mediated by an allosteric interaction of Arf6-GTP with the PH-Ct domain of EFA6 and monitors the activation of Arf1 and Arf6 differentially. These observations reveal that EFA6, BRAG, and cytohesins have unanticipated commonalities associated with divergent regulatory regimes. An important implication is that EFA6 and cytohesins may combine in a mixed negative-positive feedback loop. By allowing EFA6 to sustain a pool of dormant Arf6-GTP, such a circuit would fulfill the absolute requirement of cytohesins for activation by Arf-GTP before amplification of their GEF activity by their positive feedback loop (Padovani, 2016).
Members of the Arf family of small G proteins are involved in membrane traffic and organelle structure. They control the recruitment of coat proteins, and modulate the structure of actin filaments and the lipid composition of membranes. The ADP-ribosylation factor 6 (Arf6) isoform and the exchange factor for Arf6 (EFA6) are known to regulate the endocytic pathway of many different receptors. To determine the molecular mechanism of the EFA6/Arf6 function in vesicular transport, this study sought for new EFA6 partners. In a two-hybrid screening using the catalytic Sec7 domain as a bait, endophilin was identified as a new partner of EFA6. Endophilin contains a Bin/Amphiphysin/Rvs (BAR) domain responsible for membrane bending, and an SH3 domain responsible for the recruitment of dynamin and synaptojanin, two proteins involved, respectively, in the fission and uncoating of clathrin-coated vesicles. By using purified proteins, the direct interaction was confirmed, and the N-BAR domain was identified as the binding motif to EFA6A. Endophilin stimulates the catalytic activity of EFA6A on Arf6. In addition, it was observed that the Sec7 domain competes with flat but not with highly curved lipid membranes to bind the N-BAR. In cells, expression of EFA6A recruits endophilin to EFA6A-positive plasma membrane ruffles, whereas expression of endophilin rescues the EFA6A-mediated inhibition of transferrin internalization. Overall, these results support a model whereby EFA6 recruits endophilin on flat areas of the plasma membrane to control Arf6 activation and clathrin-mediated endocytosis (Boulakirba, 2014).
ADP ribosylation factor (Arf) 6 anchors to the plasma membrane, where it coordinates membrane trafficking and cytoskeleton remodelling, but how it assembles actin filaments is unknown. By reconstituting membrane-associated actin assembly mediated by the WASP family veroprolin homolog (WAVE) regulatory complex (WRC), this study recapitulated an Arf6-driven actin polymerization pathway. Arf6 is shown to be divergent from other Arf members, as it was incapable of directly recruiting WRC. Arf6 triggers actin assembly at the membrane indirectly by recruiting the Arf guanine nucleotide exchange factor (GEF) ARNO that activates Arf1 to enable WRC-dependent actin assembly. The pathogen Salmonella usurped Arf6 for host cell invasion by recruiting its canonical GEFs EFA6 and BRAG2. Arf6 and its GEFs facilitated membrane ruffling and pathogen invasion via ARNO, and triggered actin assembly by generating an Arf1-WRC signaling hub at the membrane in vitro and in cells. This study reconstitutes Arf6-dependent actin assembly to reveal a mechanism by which related Arf GTPases orchestrate distinct steps in the WRC cytoskeleton remodelling pathway (Humphreys, 2013).
Microtubules are polymers of tubulin heterodimers that exhibit dynamic instability: periods of growth followed by periods of shrinkage. However, the molecular regulation of dynamic instability remains elusive. This study shows that EFA-6, a cortically-localized protein, limits the growth of microtubules near the cell cortex of early embryonic cells from Caenorhabditis elegans, possibly by inducing microtubule catastrophes. Compared with wild type, embryos lacking EFA-6 had abnormally long and dense microtubules at the cell cortex, and growing microtubule plus ends resided at the cortex for up to five-fold longer. Loss of EFA-6 also caused excess centrosome separation and displacement towards the cell cortex early in mitosis, and subsequently a loss of anaphase spindle-pole oscillations and increased rates of spindle elongation. The centrosome separation phenotype was dependent on the motor protein dynein, suggesting a possible link between the modulation of microtubule dynamics at the cortex and dynein-dependent force production. EFA-6 orthologues activate ARF6-type GTPases to regulate vesicle trafficking. However, this study shows that only the C. elegans EFA-6 amino-terminus is both necessary and sufficient to limit microtubule growth along the cortex, and that this function is independent of ARF-6 (O'Rourke, 2010).
Search PubMed for articles about Drosophila Efa6
Blum, I. R., Behling-Hess, C., Padilla-Rodriguez, M., Momtaz, S., Cox, C. and Wilson, J. M. (2020). Rab22a regulates the establishment of epithelial polarity. Small GTPases: 1-12. PubMed ID: 32281471
Boulakirba, S., Macia, E., Partisani, M., Lacas-Gervais, S., Brau, F., Luton, F. and Franco, M. (2014). Arf6 exchange factor EFA6 and endophilin directly interact at the plasma membrane to control clathrin-mediated endocytosis. Proc Natl Acad Sci U S A 111(26): 9473-9478. PubMed ID: 24979773
Bu, S., Yong, W. L., Lim, B. J. W., Kondo, S., Yu, F. (2021). A systematic analysis of microtubule-destabilizing factors during dendrite pruning in Drosophila. EMBO Rep, 22(10):e52679 PubMed ID: 34338441
Chen, L., Wang, Z., Ghosh-Roy, A., Hubert, T., Yan, D., O'Rourke, S., Bowerman, B., Wu, Z., Jin, Y. and Chisholm, A. D. (2011). Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron 71(6): 1043-1057. PubMed ID: 21943602
Chen, L., Chuang, M., Koorman, T., Boxem, M., Jin, Y. and Chisholm, A. D. (2015). Axon injury triggers EFA-6 mediated destabilization of axonal microtubules via TACC and doublecortin like kinase. Elife 4. PubMed ID: 26339988
Eva, R., Koseki, H., Kanamarlapudi, V. and Fawcett, J. W. (2017). EFA6 regulates selective polarised transport and axon regeneration from the axon initial segment. J Cell Sci 130(21): 3663-3675. PubMed ID: 28935671
Fukaya, M., Ohta, S., Hara, Y., Tamaki, H. and Sakagami, H. (2016). Distinct subcellular localization of alternative splicing variants of EFA6D, a guanine nucleotide exchange factor for Arf6, in the mouse brain. J Comp Neurol 524(13): 2531-2552. PubMed ID: 27241101
Gonzalez, D. A., Jia, T., Pinzon, J. H., Acevedo, S. F., Ojelade, S. A., Xu, B., Tay, N., Desrivieres, S., Hernandez, J. L., Banaschewski, T., Buchel, C., Bokde, A. L. W., Conrod, P. J., Flor, H., Frouin, V., Gallinat, J., Garavan, H., Gowland, P. A., Heinz, A., Ittermann, B., Lathrop, M., Martinot, J. L., Paus, T., Smolka, M. N., Consortium, I., Rodan, A. R., Schumann, G. and Rothenfluh, A. (2018). The Arf6 activator Efa6/PSD3 confers regional specificity and modulates ethanol consumption in Drosophila and humans. Mol Psychiatry 23(3): 621-628. PubMed ID: 28607459
Ghosh, M., Lo, R., Ivic, I., Aguilera, B., Qendro, V., Devarakonda, C. and Shapiro, L. H. (2019). CD13 tethers the IQGAP1-ARF6-EFA6 complex to the plasma membrane to promote ARF6 activation, beta1 integrin recycling, and cell migration. Sci Signal 12(579). PubMed ID: 31040262
Hahn, I., Voelzmann, A., Liew, Y. T., Costa-Gomes, B. and Prokop, A. (2019). The model of local axon homeostasis - explaining the role and regulation of microtubule bundles in axon maintenance and pathology. Neural Development. 14:11. PubMed ID: 31706327
Humphreys, D., Davidson, A. C., Hume, P. J., Makin, L. E. and Koronakis, V. (2013). Arf6 coordinates actin assembly through the WAVE complex, a mechanism usurped by Salmonella to invade host cells. Proc Natl Acad Sci U S A 110(42): 16880-16885. PubMed ID: 24085844
Kanamarlapudi, V. (2014). Exchange factor EFA6R requires C-terminal targeting to the plasma membrane to promote cytoskeletal rearrangement through the activation of ADP-ribosylation factor 6 (ARF6). J Biol Chem 289(48): 33378-33390. PubMed ID: 25296758
Macia, E., Partisani, M., Wang, H., Lacas-Gervais, S., Le Clainche, C., Luton, F. and Franco, M. (2019). The C-terminal domain of EFA6A interacts directly with F-actin and assembles F-actin bundles. Sci Rep 9(1): 19209. PubMed ID: 31844082
O'Rourke, S. M., Christensen, S. N. and Bowerman, B. (2010). Caenorhabditis elegans EFA-6 limits microtubule growth at the cell cortex. Nat Cell Biol 12(12): 1235-1241. PubMed ID: 21076413
Padovani, D., Folly-Klan, M., Labarde, A., Boulakirba, S., Campanacci, V., Franco, M., Zeghouf, M. and Cherfils, J. (2014). EFA6 controls Arf1 and Arf6 activation through a negative feedback loop. Proc Natl Acad Sci U S A 111(34): 12378-12383. PubMed ID: 25114232
Peru, Y. C. d. P. R. L., Acevedo, S. F., Rodan, A. R., Chang, L. Y., Eaton, B. A. and Rothenfluh, A. (2012). Adult neuronal Arf6 controls ethanol-induced behavior with Arfaptin downstream of Rac1 and RhoGAP18B. J Neurosci 32(49): 17706-17713. PubMed ID: 23223291
Prokop, A. (2016). Fruit flies in biological research. Biological Sciences Review. 28:10-14
Qu, Y., Hahn, I., Lees, M., Parkin, J., Voelzmann, A., Dorey, K., Rathbone, A., Friel, C. T., Allan, V. J., Okenve-Ramos, P., Sanchez-Soriano, N. and Prokop, A. (2019). Efa6 protects axons and regulates their growth and branching by inhibiting microtubule polymerisation at the cortex. Elife 8. PubMed ID: 31718774
Yogev, S., Cooper, R., Fetter, R., Horowitz, M. and Shen, K. (2016). Microtubule organization determines axonal transport dynamics. Neuron 92(2): 449-460. PubMed ID: 27764672
date revised: August 14th 2020
Home page: The Interactive Fly © 2011 Thomas Brody, Ph.D.
{kind=link}