InteractiveFly: GeneBrief
Target of rapamycin: Biological Overview , | Regulation | Developmental Biology | Effects of Mutation | Evolutionary Homologs | References
|
Gene name - Target of rapamycin
Synonyms - dTOR, mechanistic target of rapamycin (mTOR), TORC1, FRAP/TOR Cytological map position - 34A4 Function - signaling Keywords - Tor pathway, growth, nutrient sensing |
Symbol - Tor FlyBase ID: FBgn0021796 Genetic map position - 2L Classification - Phosphatidylinositol 3- and 4-kinase Cellular location - cytoplasmic |
| Recent literature | Takats, S., Varga, A., Pircs, K. and Juhasz, G. (2015). Loss of Drosophila Vps16A enhances autophagosome formation through reduced TOR activity. Autophagy [Epub ahead of print] PubMed ID: 26061715
Summary: The HOPS tethering complex facilitates autophagosome-lysosome fusion by binding to Syntaxin 17, the autophagosomal SNARE. This study show that loss of the core HOPS complex subunit Vps16A enhances autophagosome formation and slows down Drosophila development. Mechanistically, Tor kinase is less active in Vps16A mutants likely due to impaired endocytic and biosynthetic transport to the lysosome, a site of its activation. Tor reactivation by overexpression of Rheb suppresses autophagosome formation and restores growth and developmental timing in these animals. Thus, Vps16A reduces autophagosome numbers both by indirectly restricting their formation rate and by directly promoting their clearance. In contrast, the loss of Syx17/Syntaxin 17 blocks autophagic flux without affecting the induction step in Drosophila. |
Bargiela, A., Cerro-Herreros, E., Fernandez-Costa, J. M., Vilchez, J. J., Llamusi, B. and Artero, R. (2015). Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis Model Mech 8: 679-690. PubMed ID: 26092529
Summary: Muscle mass wasting is one of the most debilitating symptoms of myotonic dystrophy type 1 (DM1) disease, ultimately leading to immobility, respiratory defects, dysarthria, dysphagia and death in advanced stages of the disease. In order to study the molecular mechanisms leading to the degenerative loss of adult muscle tissue in DM1, an inducible Drosophila model of expanded CTG trinucleotide repeat toxicity was generated that resembles an adult-onset form of the disease. Heat-shock induced expression of 480 CUG repeats in adult flies resulted in a reduction in the area of the indirect flight muscles. In these model flies, reduction of muscle area was concomitant with increased apoptosis and autophagy. Inhibition of apoptosis or autophagy mediated by the overexpression of DIAP1, mTOR (also known as Tor) or muscleblind, or by RNA interference (RNAi)-mediated silencing of autophagy regulatory genes, achieved a rescue of the muscle-loss phenotype. In fact, mTOR overexpression rescued muscle size to a size comparable to that in control flies. These results were validated in skeletal muscle biopsies from DM1 patients in which it was found downregulated autophagy and apoptosis repressor genes, and also in DM1 myoblasts where increased autophagy was found. These findings provide new insights into the signaling pathways involved in DM1 disease pathogenesis. |
Fan, X., Liang, Q., Lian, T., Wu, Q., Gaur, U., Li, D., Yang, D., Mao, X., Jin, Z., Li, Y. and Yang, M. (2015). Rapamycin preserves gut homeostasis during Drosophila aging. Oncotarget [Epub ahead of print]. PubMed ID: 26431326 Summary: Gut homeostasis plays an important role in maintaining the overall body health during aging. Rapamycin, a specific inhibitor of mTOR, exerts prolongevity effects in evolutionarily diverse species. However, its impact on the intestinal homeostasis remains poorly understood. This study demonstrates that rapamycin can slow down the proliferation rate of intestinal stem cells (ISCs) in the aging guts and induce autophagy in the intestinal epithelium in Drosophila. Rapamycin can also significantly affect the FOXO associated genes in intestine and up-regulate the negative regulators of IMD/Rel pathway, consequently delaying the microbial expansion in the aging guts. Collectively, these findings reveal that rapamycin can delay the intestinal aging by inhibiting mTOR and thus keeping stem cell proliferation in check. These results further explain the mechanism of healthspan and lifespan extension by rapamycin in Drosophila. |
Etchegaray, J. I., Elguero, E. J., Tran, J. A., Sinatra, V., Feany, M. B. and McCall, K. (2016). Defective phagocytic corpse processing results in neurodegeneration and can be rescued by TORC1 activation. J Neurosci 36: 3170-3183. PubMed ID: 26985028 Summary: The removal of apoptotic cell corpses is important for maintaining homeostasis. Previously, defects in apoptotic cell clearance have been linked to neurodegeneration. However, the mechanisms underlying this are still poorly understood. This study reports that the absence of the phagocytic receptor Draper in glia leads to a pronounced accumulation of apoptotic neurons in the brain of Drosophila melanogaster. These dead cells persist in the brain throughout the lifespan of the organism and are associated with age-dependent neurodegeneration. The data indicate that corpses persist because of defective phagosome maturation, rather than recognition defects. TORC1 activation, or inhibition of Atg1, in glia is sufficient to rescue corpse accumulation as well as neurodegeneration. These results suggest that phagocytosis of apoptotic neurons by glia during development is essential for brain homeostasis in adult flies. Furthermore, it suggests that TORC1 regulates Draper-mediated phagosome maturation. Previously, defects in dead cell clearance were linked to neurodegeneration, but the exact mechanisms are not well understood. This study reports that the absence of an engulfment receptor leads to a pronounced accumulation of dead neurons in the brain of the fruit fly Drosophila melanogaster. These dead cells persist in the brain throughout the lifespan of the organism and are associated with age-dependent neurodegeneration. The data indicate that corpses persist because of defective degradation of cells rather than recognition of dead cells. |
Liu, D., Shaukat, Z., Xu, T., Denton, D., Saint, R. and Gregory, S. (2016). Autophagy regulates the survival of cells with chromosomal instability. Oncotarget [Epub ahead of print]. PubMed ID: 27590505
Summary: Chromosomal instability (CIN) refers to genomic instability in which cells have gained or lost chromosomes or chromosomal fragments. A high level of CIN is common in solid tumours and is associated with cancer drug resistance and poor prognosis. The impact of CIN-induced stress and the resulting cellular responses are only just beginning to emerge. Using proliferating tissue in Drosophila as a model, this study found that autophagy is activated in CIN cells and is necessary for their survival. Specifically, increasing the removal of defective mitochondria by mitophagy is able to lower levels of reactive oxygen species and the resultant cellular damage that is normally seen in CIN cells. In response to DNA damage, CIN is increased in a positive feedback loop, and increasing autophagy by Tor depletion was found to decrease the level of CIN in proliferating cells. These findings underline the importance of autophagy control in the development of CIN tumours. |
Braden, C. R. and Neufeld, T. P. (2016). Atg1-independent induction of autophagy by the Drosophila Ulk3 homolog ADUK. FEBS J [Epub ahead of print]. PubMed ID: 27717182
Summary: Although canonical autophagy regulation requires a multi-protein complex centered on the Ser/Thr-kinase Atg1 (mammalian Ulk1/2), alternative signals can induce autophagy independent of Atg1 through unknown mechanisms. This study identified the Drosophila Ulk3 ortholog, another Drosophila Unc-51-like kinase (ADUK: CG8866), as an Atg1-independent autophagy inducer. ADUK interacts with Atg1 complex members Atg13 and 200 kDa FAK family kinase-interacting protein (FIP200 or Atg17), and requires Atg13 but not Atg1 for autophagy induction. Loss of ADUK shortens adult lifespan and reduces the autophagic response to a chemical stressor, dimethyl sulfoxide. However, ADUK is not required for autophagy induction by Atg1-dependent nutrient or developmental cues. Atg1 and ADUK/Ulk3 thus represent alternative catalytic components of a shared autophagy kinase complex. |
Ohhara, Y., Kobayashi, S. and Yamanaka, N.
(2017). Nutrient-dependent
endocycling in steroidogenic tissue dictates timing of metamorphosis in
Drosophila melanogaster. PLoS Genet 13: e1006583. PubMed
ID: 28121986 Summary: Many animals have an intrinsic growth checkpoint during juvenile development, after which an irreversible decision is made to upregulate steroidogenesis, triggering the metamorphic juvenile-to-adult transition. However, a molecular process underlying such a critical developmental decision remains obscure. This study shows that nutrient-dependent endocycling in steroidogenic cells provides the machinery necessary for irreversible activation of metamorphosis in Drosophila melanogaster. Endocycle progression in cells of the prothoracic gland (PG) is tightly coupled with the growth checkpoint, and block of endocycle in PG cells causes larval developmental arrest due to reduction in biosynthesis of the steroid hormone ecdysone. Moreover, inhibition of the nutrient sensor target of rapamycin (TOR) in the PG during the checkpoint period causes endocycle inhibition and developmental arrest, which can be rescued by inducing additional rounds of endocycles by Cyclin E. The study proposes that a TOR-mediated cell cycle checkpoint in steroidogenic tissue provides a systemic growth checkpoint for reproductive maturation. |
Yoon, W. H., Sandoval, H., Nagarkar-Jaiswal, S., Jaiswal, M., Yamamoto, S., Haelterman, N. A., Putluri, N., Putluri, V., Sreekumar, A., Tos, T., Aksoy, A., Donti, T., Graham, B. H., Ohno, M., Nishi, E., Hunter, J., Muzny, D. M., Carmichael, J., Shen, J., Arboleda, V. A., Nelson, S. F., Wangler, M. F., Karaca, E., Lupski, J. R. and Bellen, H. J. (2016). Loss of Nardilysin, a mitochondrial co-chaperone for alpha-Ketoglutarate dehydrogenase, promotes mTORC1 activation and neurodegeneration. Neuron [Epub ahead of print]. PubMed ID: 28017472
Summary: Mutations in Nardilysin (dNrd1) were identified in a forward genetic screen designed to isolate genes whose loss causes neurodegeneration in Drosophila photoreceptor neurons. NRD1 is localized to mitochondria, where it recruits mitochondrial chaperones and assists in the folding of alpha-ketoglutarate dehydrogenase (OGDH), a rate-limiting enzyme in the Krebs cycle. Loss of Nrd1 or Ogdh leads to an increase in alpha-ketoglutarate, a substrate for OGDH, which in turn leads to mTORC1 activation and a subsequent reduction in autophagy. Inhibition of mTOR activity by rapamycin or partially restoring autophagy delays neurodegeneration in dNrd1 mutant flies. In summary, this study reveals a novel role for NRD1 as a mitochondrial co-chaperone for OGDH and provides a mechanistic link between mitochondrial metabolic dysfunction, mTORC1 signaling, and impaired autophagy in neurodegeneration. |
Tang, H. W., Hu, Y., Chen, C. L., Xia, B., Zirin, J., Yuan, M., Asara, J. M., Rabinow, L. and Perrimon, N. (2018). The TORC1-regulated CPA complex rewires an RNA processing network to drive autophagy and metabolic reprogramming. Cell Metab 27(5): 1040-1054.e1048. PubMed ID: 29606597
Summary: Nutrient deprivation induces autophagy through inhibiting TORC1 activity. This study describes a novel mechanism in Drosophila by which TORC1 regulates RNA processing of Atg transcripts and alters ATG protein levels and activities via the cleavage and polyadenylation (CPA) complex. TORC1 signaling inhibits CDK8 and DOA kinases, which directly phosphorylate CPSF6, a component of the CPA complex. These phosphorylation events regulate CPSF6 localization, RNA binding, and starvation-induced alternative RNA processing of transcripts involved in autophagy, nutrient, and energy metabolism, thereby controlling autophagosome formation and metabolism. Similarly, it was found that mammalian CDK8 and CLK2, a DOA ortholog, phosphorylate CPSF6 to regulate autophagy and metabolic changes upon starvation, revealing an evolutionarily conserved mechanism linking TORC1 signaling with RNA processing, autophagy, and metabolism. |
Cara, F. D., Bulow, M., Simmonds, A. J. and Rachubinski, R. A. (2018). Dysfunctional peroxisomes compromise gut structure and host defense by increased cell death and Tor-dependent autophagy. Mol Biol Cell: mbcE18070434. PubMed ID: 30188767
Summary: The gut has a central role in digestion and nutrient absorption, but it also serves in defending against pathogens, engages in mutually beneficial interactions with commensals, and is a major source of endocrine signals. Gut homeostasis is necessary for organismal health, and changes to the gut are associated with conditions like obesity and diabetes and inflammatory illnesses like Crohn's disease. This study reports that peroxisomes, organelles involved in lipid metabolism and redox balance, are required to maintain gut epithelium homeostasis and renewal in Drosophila and for survival and development of the organism. Dysfunctional peroxisomes in gut epithelial cells activate Tor kinase-dependent autophagy that increases cell death and epithelial instability, which ultimately alter the composition of the intestinal microbiota, compromise immune pathways in the gut in response to infection, and affect organismal survival. Peroxisomes in the gut effectively function as hubs that coordinate responses from stress, metabolic and immune signaling pathways to maintain enteric health and the functionality of the gut-microbe interface. |
Kim, A. R. and Choi, K. W. (2019). TRiC/CCT chaperonins are essential for organ growth by interacting with insulin/TOR signaling in Drosophila. Oncogene. PubMed ID: 30792539
Summary: Organ size is regulated by intercellular signaling for cell growth and proliferation. The TOR pathway mediates a key signaling mechanism for controlling cell size and number in organ growth. Chaperonin containing TCP-1 (CCT) is a complex that assists protein folding and function, but its role in animal development is largely unknown. This study shows that the CCT complex is required for organ growth by interacting with the TOR pathway in Drosophila. Reduction of CCT4 results in growth defects by affecting both cell size and proliferation. Loss of CCT4 causes preferential cell death anterior to the morphogenetic furrow in the eye disc and within wing pouch in the wing disc. Depletion of any CCT subunit in the eye disc results in headless phenotype. Overgrowth by active TOR signaling is suppressed by CCT RNAi. The CCT complex physically interacts with TOR signaling components including TOR, Rheb, and S6K. Loss of CCT leads to decreased phosphorylation of S6K and S6 while increasing phosphorylation of Akt. Insulin/TOR signaling is also necessary and sufficient for promoting CCT complex transcription. These data provide evidence that the CCT complex regulates organ growth by directly interacting with the TOR signaling pathway. |
Lee, B., Barretto, E. C. and Grewal, S. S. (2019). TORC1 modulation in adipose tissue is required for organismal adaptation to hypoxia in Drosophila. Nat Commun 10(1): 1878. PubMed ID: 31015407
Summary: Animals often develop in environments where conditions such as food, oxygen and temperature fluctuate. The ability to adapt their metabolism to these fluctuations is important for normal development and viability. In most animals, low oxygen (hypoxia) is deleterious. However some animals can alter their physiology to tolerate hypoxia. This study shows that TORC1 modulation in adipose tissue is required for organismal adaptation to hypoxia in Drosophila. Hypoxia rapidly suppresses TORC1 signaling in Drosophila larvae via TSC-mediated inhibition of Rheb. This hypoxia-mediated inhibition of TORC1 specifically in the larval fat body is essential for viability. Moreover, these effects of TORC1 inhibition on hypoxia tolerance are mediated through remodeling of fat body lipid storage. These studies identify the larval adipose tissue as a key hypoxia-sensing tissue that coordinates whole-body development and survival to changes in environmental oxygen by modulating TORC1 and lipid metabolism. |
Ramanathan, S. P., Krajnc, M. and Gibson, M. C. (2019). Cell-size pleomorphism drives aberrant clone dispersal in proliferating epithelia. Dev Cell. PubMed ID: 31495693
Summary: As epithelial tissues develop, groups of cells related by descent tend to associate in clonal populations rather than dispersing within the cell layer. While this is frequently assumed to be a result of differential adhesion, precise mechanisms controlling clonal cohesiveness remain unknown. This study employed computational simulations to modulate epithelial cell size in silico and shows that junctions between small cells frequently collapse, resulting in clone-cell dispersal among larger neighbors. Consistent with similar dynamics in vivo, it was further demonstrated that mosaic disruption of Drosophila Tor generates small cells and results in aberrant clone dispersal in developing wing disc epithelia. A geometric basis is proposed for this phenomenon, supported in part by the observation that soap-foam cells exhibit similar size-dependent junctional rearrangements. Combined, these results establish a link between cell-size pleomorphism and the control of epithelial cell packing, with potential implications for understanding tumor cell dispersal in human disease. |
Potter, S., Sifers, J., Yocom, E., Blumich, S. L. E., Potter, R., Nadolski, J., Harrison, D. A. and Cooper, R. L. (2019). Effects of inhibiting mTOR with rapamycin on behavior, development, neuromuscular physiology and cardiac function in larval Drosophila. Biol Open 8(11). PubMed ID: 31704693
Summary: Rapamycin and other mTOR inhibitors are being heralded as possible treatments for many human ailments. It is currently being utilized clinically as an immunomodulator after transplantation procedures and as a treatment for certain forms of cancer, but it has numerous potential clinical indications. Some studies have shown profound effects on life cycle and muscle physiology, but these issues have not been addressed in an organism undergoing developmental processes. This paper fills this void by examining the effect of mTOR inhibition by rapamycin on several different qualities of larval Drosophila. Various dosages of the compound were fed to second instar larvae. These larvae were monitored for pupae formation to elucidate possible life cycle effects, and a delay to pupation was quantified. Behavioral deficits were documented in rapamycin-treated larvae. Electrophysiological measurements were taken to discern changes in muscle physiology and synaptic signaling (i.e. resting membrane potential, amplitude of excitatory post-synaptic potentials, synaptic facilitation). Pupation delay and effects on behavior that are likely due to synaptic alterations within the central nervous system were discovered in rapamycin-fed larvae. These results allow for several conclusions as to how mTOR inhibition by rapamycin affects a developing organism. This could eventually allow for a more informed decision when using rapamycin and other mTOR inhibitors to treat human diseases, especially in children and adolescents, to account for known side effects. |
Li, Y., Romey-Glusing, R., Tahan Zadeh, N., von Frieling, J., Hoffmann, J., Huebbe, P., Bruchhaus, I., Rimbach, G., Fink, C. and Roeder, T. (2020). Furbellow (Brown Algae) Extract Increases Lifespan in Drosophila by Interfering with TOR-Signaling. Nutrients 12(4). PubMed ID: 32331413
Summary: Algal products are well known for their health promoting effects. Nonetheless, an in depth understanding of the underlying molecular mechanisms is still only fragmentary. This study shows that aqueous furbelow extracts (brown algae, Saccorhiza polyschides) lengthen the life of both sexes of the fruit fly Drosophila melanogaster substantially, if used as nutritional additives to conventional food. This life prolonging effect became even more pronounced in the presence of stressors, such as high-fat dieting of living under drought conditions. Application of the extracts did not change food intake, excretion, or other major physiological parameters. Nevertheless, effects on the intestinal microbiota were observed, leading to an increased species richness, which is usually associated with healthy conditions. Lifespan extension was not observed in target of rapamycin (TOR)-deficient animals, implying that functional TOR signaling is necessary to unfold the positive effects of brown algae extract (BAE) on this important trait. The lack of life lengthening in animals with deregulated TOR signaling exclusively targeted to body fat showed that this major energy storage organ is instrumental for transmitting these effects. In addition, expression of Imaginal morphogenesis protein-Late 2 (Imp-L2), an effective inhibitor of insulin signaling implies that BAE exerts their positive effects through interaction with the tightly interwoven TOR- and insulin-signaling systems, although insulin levels were not directly affected by this intervention. |
Recasens-Alvarez, C., Alexandre, C., Kirkpatrick, J., Nojima, H., Huels, D. J., Snijders, A. P. and Vincent, J. P. (2021). Ribosomopathy-associated mutations cause proteotoxic stress that is alleviated by TOR inhibition. Nat Cell Biol 23(2): 127-135. PubMed ID: 33495632
Summary: Ribosomes are multicomponent molecular machines that synthesize all of the proteins of living cells. Most of the genes that encode the protein components of ribosomes are therefore essential. A reduction in gene dosage is often viable albeit deleterious and is associated with human syndromes, which are collectively known as ribosomopathies. The cell biological basis of these pathologies has remained unclear. This study modelled human ribosomopathies in Drosophila and found widespread apoptosis and cellular stress in the resulting animals. This is not caused by insufficient protein synthesis, as reasonably expected. Instead, ribosomal protein deficiency elicits proteotoxic stress, which is suggested to be caused by the accumulation of misfolded proteins that overwhelm the protein degradation machinery. Dampening the integrated stress response or autophagy increases the harm inflicted by ribosomal protein deficiency, suggesting that these activities could be cytoprotective. Inhibition of TOR activity-which decreases ribosomal protein production, slows down protein synthesis and stimulates autophagy-reduces proteotoxic stress in the ribosomopathy model. Interventions that stimulate autophagy, combined with means of boosting protein quality control, could form the basis of a therapeutic strategy for this class of diseases. |
Santiago, J. C., Boylan, J. M., Lemieux, F. A., Gruppuso, P. A., Sanders, J. A. and Rand, D. M. (2021). Mitochondrial genotype alters the impact of rapamycin on the transcriptional response to nutrients in Drosophila. BMC Genomics 22(1): 213. PubMed ID: 33761878
Summary: In addition to their well characterized role in cellular energy production, new evidence has revealed the involvement of mitochondria in diverse signaling pathways that regulate a broad array of cellular functions. The mitochondrial genome (mtDNA) encodes essential components of the oxidative phosphorylation (OXPHOS) pathway whose expression must be coordinated with the components transcribed from the nuclear genome. Mitochondrial dysfunction is associated with disorders including cancer and neurodegenerative diseases, yet the role of the complex interactions between the mitochondrial and nuclear genomes are poorly understood. Using a Drosophila model in which alternative mtDNAs are present on a common nuclear background, the effects of this altered mitonuclear communication on the transcriptomic response to altered nutrient status were studied. Adult flies with the 'native' and 'disrupted' genotypes were re-fed following brief starvation, with or without exposure to rapamycin, the cognate inhibitor of the nutrient-sensing target of rapamycin (TOR). RNAseq showed that alternative mtDNA genotypes affect the temporal transcriptional response to nutrients in a rapamycin-dependent manner. Pathways most greatly affected were OXPHOS, protein metabolism and fatty acid metabolism. A distinct set of testis-specific genes was also differentially regulated in the experiment. It is concluded that any of the differentially expressed genes between alternative mitonuclear genotypes have no direct interaction with mtDNA gene products, suggesting that the mtDNA genotype contributes to retrograde signaling from mitochondria to the nucleus. The interaction of mitochondrial genotype (mtDNA) with rapamycin treatment identifies new links between mitochondria and the nutrient-sensing mTORC1 (mechanistic target of rapamycin complex 1) signaling pathway. |
Kohashi, K., Mori, Y., Narumi, R., Kozawa, K., Kamasaki, T., Ishikawa, S., Kajita, M., Kobayashi, R., Tamori, Y. and Fujita, Y. (2021). Sequential oncogenic mutations influence cell competition. Curr Biol. PubMed ID: 34314674
Summary: At the initial stage of carcinogenesis, newly emerging transformed cells are often eliminated from epithelial layers via cell competition with the surrounding normal cells. For instance, when surrounded by normal cells, oncoprotein RasV12-transformed cells are extruded into the apical lumen of epithelia. During cancer development, multiple oncogenic mutations accumulate within epithelial tissues. However, it remains elusive whether and how cell competition is also involved in this process. Using a mammalian cell culture model system, this study investigated what happens upon the consecutive mutations of Ras and tumor suppressor protein Scribble. When Ras mutation occurs under the Scribble-knockdown background, apical extrusion of Scribble/Ras double-mutant cells is strongly diminished. In addition, at the boundary with Scribble/Ras cells, Scribble-knockdown cells frequently undergo apoptosis and are actively engulfed by the neighboring Scribble/Ras cells. The comparable apoptosis and engulfment phenotypes are also observed in Drosophila epithelial tissues between Scribble/Ras double-mutant and Scribble single-mutant cells. Furthermore, mitochondrial membrane potential is enhanced in Scribble/Ras cells, causing the increased mitochondrial reactive oxygen species (ROS). Suppression of mitochondrial membrane potential or ROS production diminishes apoptosis and engulfment of the surrounding Scribble-knockdown cells, indicating that mitochondrial metabolism plays a key role in the competitive interaction between double- and single-mutant cells. Moreover, mTOR (mechanistic target of rapamycin kinase) acts downstream of these processes. These results imply that sequential oncogenic mutations can profoundly influence cell competition, a transition from loser to winner. Further studies would open new avenues for cell competition-based cancer treatment, thereby blocking clonal expansion of more malignant populations within tumors. |
Szlachcic, E. and Czarnoleski, M. (2021). Thermal and Oxygen Flight Sensitivity in Ageing Drosophila melanogaster Flies: Links to Rapamycin-Induced Cell Size Changes. Biology (Basel) 10(9). PubMed ID: 34571738
Summary: Ectotherms can become physiologically challenged when performing oxygen-demanding activities (e.g., flight) across differing environmental conditions, specifically temperature and oxygen levels. Achieving a balance between oxygen supply and demand can also depend on the cellular composition of organs, which either evolves or changes plastically in nature; however, this hypothesis has rarely been examined, especially in tracheated flying insects. The relatively large cell membrane area of small cells should increase the rates of oxygen and nutrient fluxes in cells; however, it does also increase the costs of cell membrane maintenance. To address the effects of cell size on flying insects, the wing-beat frequency was measured in two cell-size phenotypes of Drosophila melanogaster when flies were exposed to two temperatures (warm/hot) combined with two oxygen conditions (normoxia/hypoxia). The cell-size phenotypes were induced by rearing 15 isolines on either standard food (large cells) or rapamycin-enriched food (small cells). Rapamycin supplementation (downregulation of TOR activity) produced smaller flies with smaller wing epidermal cells. Flies generally flapped their wings at a slower rate in cooler (warm treatment) and less-oxygenated (hypoxia) conditions, but the small-cell-phenotype flies were less prone to oxygen limitation than the large-cell-phenotype flies and did not respond to the different oxygen conditions under the warm treatment. It is suggested that ectotherms with small-cell life strategies can maintain physiologically demanding activities (e.g., flight) when challenged by oxygen-poor conditions, but this advantage may depend on the correspondence among body temperatures, acclimation temperatures and physiological thermal limits. |
Sujkowski, A. and Wessells, R. (2021). Exercise and Sestrin Mediate Speed and Lysosomal Activity in Drosophila by Partially Overlapping Mechanisms. Cells 10(9). PubMed ID: 34572128
Summary: Chronic exercise is widely recognized as an important contributor to healthspan in humans and in diverse animal models. Sestrins, a family of evolutionarily conserved exercise-inducible proteins, are critical mediators of exercise benefits in flies and mice. Knockout of Sestrins prevents exercise adaptations to endurance and flight in Drosophila, and similarly prevents benefits to endurance and metabolism in exercising mice. In contrast, overexpression of dSestrin in muscle mimics several of the molecular and physiological adaptations characteristic of endurance exercise. This study extends those observations to examine the impact of dSestrin on preserving speed and increasing lysosomal activity. dSestrin was found to be a critical factor driving exercise adaptations to climbing speed, but is not absolutely required for exercise to increase lysosomal activity in Drosophila. The role of Sestrin in increasing speed during chronic exercise requires both the TORC2/AKT axis and the PGC1α homolog spargel, while dSestrin requires interactions with TORC1 to cell-autonomously increase lysosomal activity. These results highlight the conserved role of Sestrins as key factors that drive diverse physiological adaptations conferred by chronic exercise. |
Basu, I., Bar, S., Prasad, M. and Datta, R. (2022). Adipose deficiency and aberrant autophagy in a Drosophila model of MPS VII is corrected by pharmacological stimulators of mTOR. Biochim Biophys Acta Mol Basis Dis 1868(7): 166399. PubMed ID: 35318126
Summary: Mucopolysaccharidosis type VII (MPS VII) is a recessively inherited lysosomal storage disorder caused due to β-glucuronidase (β-GUS) enzyme deficiency. Prominent clinical symptoms include hydrops fetalis, musculoskeletal deformities, neurodegeneration and hepatosplenomegaly leading to premature death in most cases. Apart from these, MPS VII is also characterized as adipose storage deficiency disorder although the underlying mechanism of this lean phenotype in the patients or β-GUS-deficient mice still remains a mystery. This issue was addressed using a recently developed Drosophila model of MPS VII (the CG2135-/- fly), which also exhibited a significant loss of body fat. This study reports that the lean phenotype of the CG2135-/- larvae is due to fewer number of adipocytes, smaller lipid droplets and reduced adipogenesis. The data further revealed that there is an abnormal accumulation of autophagosomes in the CG2135-/- larvae due to autophagosome-lysosome fusion defect. Decreased lysosome-mediated turnover also led to attenuated mTOR activity in the CG2135-/- larvae. Interestingly, treatment of the CG2135-/- larvae with mTOR stimulators, 3BDO or glucose, led to the restoration of mTOR activity with simultaneous correction of the autophagy defect and adipose storage deficiency. These finding thus established a hitherto unknown mechanistic link between autophagy dysfunction, mTOR downregulation and reduced adiposity in MPS VII. |
Kakanj, P., Bhide, S., Moussian, B. and Leptin, M. (2022). Autophagy-mediated plasma membrane removal promotes the formation of epithelial syncytia. Embo j: e109992. PubMed ID: 35262206
Summary: Epithelial wound healing in Drosophila involves the formation of multinucleate cells surrounding the wound. This study shows that autophagy, a cellular degradation process often deployed in stress responses, is required for the formation of a multinucleated syncytium during wound healing, and that autophagosomes that appear near the wound edge acquire plasma membrane markers. In addition, uncontrolled autophagy in the unwounded epidermis leads to the degradation of endo-membranes and the lateral plasma membrane, while apical and basal membranes and epithelial barrier function remain intact. Proper functioning of TORC1 is needed to prevent destruction of the larval epidermis by autophagy, in a process that depends on phagophore initiation and expansion but does not require autophagosomes fusion with lysosomes. Autophagy induction can also affect other sub-cellular membranes, as shown by its suppression of experimentally induced laminopathy-like nuclear defects. These findings reveal a function for TORC1-mediated regulation of autophagy in maintaining membrane integrity and homeostasis in the epidermis and during wound healing. |
Francis, D., Burguete, A. S. and Ghabrial, A. (2022). Regulation of Archease by the mTOR-vATPase axis. Development. PubMed ID: 36111596
Summary: Larval terminal cells of the Drosophila tracheal system generate extensive branched tubes, requiring a huge increase in apical membrane. Terminal cells were compromised for apical membrane expansion - mTOR-vATPase axis and apical polarity mutants - were invaded by the neighboring stalk cell. The invading cell grows and branches, replacing the original single intercellular junction between stalk and terminal cell with multiple intercellular junctions. This study characterize disjointed, a mutation in the same phenotypic class. disjointed encodes Drosophila Archease, required for RNA ligase (RtcB) function critical for tRNA maturation and ER stress-regulated nonconventional splicing of Xbp1 mRNA. The steady-state subcellular localization of Archease is principally nuclear, and dependent upon TOR-vATPase activity. In tracheal cells mutant for Rheb or vATPase, Archease localization shifted dramatically from nucleus to cytoplasm. Further, this study found that blocking tRNA maturation by knockdown of tRNAse z also induced compensatory branching. Taken together, these data suggest that the TOR-vATPase axis promotes apical membrane growth in part through nuclear localization of Archease, where Archease is required for tRNA maturation. |
Borkowsky, S., Gass, M., Alavizargar, A., Hanewinkel, J., Hallstein, I., Nedvetsky, P., Heuer, A. and Krahn, M. P. (2023). Phosphorylation of LKB1 by PDK1 Inhibits Cell Proliferation and Organ Growth by Decreased Activation of AMPK. Cells 12(5). PubMed ID: 36899949
Summary: The master kinase LKB1 is a key regulator of several cellular processes, including cell proliferation, cell polarity and cellular metabolism. It phosphorylates and activates several downstream kinases, including AMP-dependent kinase, AMPK. Activation of AMPK by low energy supply and phosphorylation of LKB1 results in an inhibition of mTOR, thus decreasing energy-consuming processes, in particular translation and, thus, cell growth. LKB1 itself is a constitutively active kinase, which is regulated by posttranslational modifications and direct binding to phospholipids of the plasma membrane. This study reports that LKB1 binds to Phosphoinositide-dependent kinase (PDK1) by a conserved binding motif. Furthermore, a PDK1-consensus motif is located within the kinase domain of LKB1 and LKB1 gets phosphorylated by PDK1 in vitro. In Drosophila, knockin of phosphorylation-deficient LKB1 results in normal survival of the flies, but an increased activation of LKB1, whereas a phospho-mimetic LKB1 variant displays decreased AMPK activation. As a functional consequence, cell growth as well as organism size is decreased in phosphorylation-deficient LKB1. Molecular dynamics simulations of PDK1-mediated LKB1 phosphorylation revealed changes in the ATP binding pocket, suggesting a conformational change upon phosphorylation, which in turn can alter LKB1's kinase activity. Thus, phosphorylation of LKB1 by PDK1 results in an inhibition of LKB1, decreased activation of AMPK and enhanced cell growth. |
Fulton, T. L., Mirth, C. K. and Piper, M. D. W. (2022). Restricting a single amino acid cross-protects Drosophila melanogaster from nicotine poisoning through mTORC1 and GCN2 signalling. Open Biol 12(12): 220319. PubMed ID: 36514979
Summary: Dietary interventions that restrict protein intake have repeatedly been shown to offer beneficial health outcomes to the consumer. Benefits such as increased stress tolerance can be observed when individual amino acids are restricted, thus mimicking dietary protein restriction. This study sought to further understand the relationship between dietary amino acids and stress tolerance using Drosophila melanogaster. Using a chemically defined medium for Drosophila, this study found that transiently restricting adult flies of a single essential amino acid generally protects against a lethal dose of the naturally occurring insecticide, nicotine. This protection varied with the identity of the focal amino acid and depended on the duration and intensity of its restriction. To understand the molecular basis of these effects, the signalling of two cellular sensors of amino acids, GCN2 and mTORC1 were modified, in combination with amino acid restriction. GCN2 was necessary for diets to protect against nicotine, whereas the suppression of mTORC1 was sufficient to induce nicotine resistance. This finding implies that amino acid restriction acts via amino acid signalling to cross-protect against seemingly unrelated stressors. Altogether, this study offers new insights into the physiological responses to restriction of individual amino acids that confer stress tolerance. |
Gui, J., Samuels, T. J., Grobicki, K. Z. A. and Teixeira, F. K. (2023). Simultaneous activation of Tor and suppression of ribosome biogenesis by TRIM-NHL proteins promotes terminal differentiation. Cell Rep 42(3): 112181. PubMed ID: 36870055
Summary: Tissue development and homeostasis depend on the balance between growth and terminal differentiation, but the mechanisms coordinating these processes remain elusive. Accumulating evidence indicates that ribosome biogenesis (RiBi) and protein synthesis, two cellular processes sustaining growth, are tightly regulated and yet can be uncoupled during stem cell differentiation. Using the Drosophila adult female germline stem cell and larval neuroblast systems, this study showed that Mei-P26 and Brat, two Drosophila TRIM-NHL paralogs, are responsible for uncoupling RiBi and protein synthesis during differentiation. In differentiating cells, Mei-P26 and Brat activate the target of rapamycin (Tor) kinase to promote translation, while concomitantly repressing RiBi. Depletion of Mei-P26 or Brat results in defective terminal differentiation, which can be rescued by ectopic activation of Tor together with suppression of RiBi. These results indicate that uncoupling RiBi and translation activities by TRIM-NHL activity creates the conditions required for terminal differentiation. |
Rodrigues, N. R., Macedo, G. E., Martins, I. K., Vieira, P. B., Kich, K. G., Posser, T. and Franco, J. L. (2023). Sleep disturbance induces a modulation of clock gene expression and alters metabolism regulation in Drosophila. Physiol Behav: 114334. PubMed ID: 37595818
Summary: Sleep disorders are catching attention worldwide as they can induce dyshomeostasis and health issues in all animals, including humans. Circadian rhythms are biological 24-hour cycles that influence physiology and behavior in all living organisms. Sleep is a crucial resting state for survival and is under the control of circadian rhythms. Studies have shown the influence of sleep on various pathological conditions, including metabolic diseases; however, the biological mechanisms involving the circadian clock, sleep, and metabolism regulation are not well understood. Previous work standardized a sleep disturbance protocol and, observed that short-time sleep deprivation and sleep-pattern alteration induce homeostatic sleep regulation, locomotor deficits, and increase oxidative stress. This study investigated the relationship between these alterations with the circadian clock and energetic metabolism. This study evaluated the expression of the circadian clock and Drosophila insulin-like peptides (DILPs) genes and metabolic markers glucose, triglycerides, and glycogen in fruit flies subjected to short-term sleep disruption protocols. The sleep disturbance altered the expression of clock genes and DILP gene expression, and modulated glucose, triglycerides, and glycogen levels. Moreover, changes were demonstrated in mTor/dFoxo genes, AKT phosphorylation, and dopamine levels in nocturnal light-exposed flies. Thus, these results suggest a connection between clock genes and metabolism disruption as a consequence of sleep disruption, demonstrating the importance of sleep quality in health maintenance. |
Srivastav, S., van der Graaf, K., Singh, P., Utama, A. B., Meyer, M. D., McNew, J. A., Stern, M. (2024). Atl (atlastin) regulates mTor signaling and autophagy in Drosophila muscle through alteration of the lysosomal network. Autophagy, 20(1):131-150 PubMed ID: 37649246
Summary: The hereditary spastic paraplegias (HSPs) represent a family of genetic disorders comprising at least 72 different genes with the common pathology of progressive locomotor deficits and spasticity. ATL1/SPG3A (atlastin GTPase 1) encodes an ER fusion protein that controls ER morphology, which implicates ER structure as a causal factor in HSP. This study used Drosophila to study effects of decreased atl (atlastin) on properties of the larval body wall muscle. We found that muscle atl loss causes accumulation of aggregates containing polyubiquitin (polyUB), mostly bound to the autophagy receptor ref(2)P/SQSTM1/p62. Muscle atl loss also decreased volume and complexity of the endolysosomal network and decreased lysosome number. To determine effects of these lysosomal deficits on progression through the basal autophagy pathway, Atg8a tagged with both GFP and mCherry in a wild-type and atl mutant background. Numerous structures containing mCherry were found but not GFP fluorescence in wild type, indicating that Atg8a was found mostly in mature autolysosomes. In contrast, muscles lacking atl exhibited significant amounts of GFP signal, indicating failure of autophagosome maturation with acidic lysosomes. Many of these GFP-positive puncta contained the late-endosome marker Rab7 but not Lamp1, indicating that some autophagy cargo was accumulating within amphisomes. It was also found that this autophagy block was accompanied by an inability to activate the mTor kinase. These results provide mechanistic insights into the role of atl in maintaining proper function of the autophagy pathway and suggests that certain pathologies in patients with mutations in ATL1/SPG3A might result from altered MTOR signaling. |
Juarez-Carreno, S., Geissmann, F. (2023). The macrophage genetic cassette inr/dtor/pvf2 is a nutritional status checkpoint for developmental timing. Sci Adv, 9(38):eadh0589 PubMed ID: 37729406
Summary: A small number of signaling molecules, used reiteratively, control differentiation programs, but the mechanisms that adapt developmental timing to environmental cues are less understood. This study reports that a macrophage inr/dtor/pvf2 genetic cassette is a developmental timing checkpoint in Drosophila, which either licenses or delays biosynthesis of the steroid hormone in the endocrine gland and metamorphosis according to the larval nutritional status. Insulin receptor/dTor signaling in macrophages is required and sufficient for production of the PDGF/VEGF family growth factor Pvf2, which turns on transcription of the sterol biosynthesis Halloween genes in the prothoracic gland via its receptor Pvr. In response to a starvation event or genetic manipulation, low Pvf2 signal delays steroid biosynthesis until it becomes Pvr-independent, thereby prolonging larval growth before pupariation. The significance of this developmental timing checkpoint for host fitness is illustrated by the observation that it regulates the size of the pupae and adult flies. |
Breznak, S. M., Peng, Y., Deng, L., Kotb, N. M., Flamholz, Z., Rapisarda, I. T., Martin, E. T., LaBarge, K. A., Fabris, D., Gavis, E. R. and Rangan, P. (2023). H/ACA snRNP-dependent ribosome biogenesis regulates translation of polyglutamine proteins. Sci Adv 9(25): eade5492. PubMed ID: 37343092
Summary: Stem cells in many systems, including Drosophila germline stem cells (GSCs), increase ribosome biogenesis and translation during terminal differentiation. This study showns that the H/ACA small nuclear ribonucleoprotein (snRNP) complex that promotes pseudouridylation of ribosomal RNA (rRNA) and ribosome biogenesis is required for oocyte specification. Reducing ribosome levels during differentiation decreased the translation of a subset of messenger RNAs that are enriched for CAG trinucleotide repeats and encode polyglutamine-containing proteins, including differentiation factors such as RNA-binding Fox protein 1. Moreover, ribosomes were enriched at CAG repeats within transcripts during oogenesis. Increasing target of rapamycin (TOR) activity to elevate ribosome levels in H/ACA snRNP complex-depleted germlines suppressed the GSC differentiation defects, whereas germlines treated with the TOR inhibitor rapamycin had reduced levels of polyglutamine-containing proteins. Thus, ribosome biogenesis and ribosome levels can control stem cell differentiation via selective translation of CAG repeat-containing transcripts. |
Willot, Q., du Toit, A., de Wet, S., Huisamen, E. J., Loos, B., Terblanche, J. S. (2023). Exploring the connection between autophagy and heat-stress tolerance in Drosophila melanogaster. Proceedings Biological sciences, 290(2006):20231305 PubMed ID: 37700658
Summary: Mechanisms aimed at recovering from heat-induced damages are closely associated with the ability of ectotherms to survive exposure to stressful temperatures. Autophagy, a ubiquitous stress-responsive catabolic process, has recently gained renewed attention as one of these mechanisms. By increasing the turnover of cellular structures as well as the clearance of long-lived protein and protein aggregates, the induction of autophagy has been linked to increased tolerance to a range of abiotic stressors in diverse ectothermic organisms. However, whether a link between autophagy and heat-tolerance exists in insect models remains unclear despite broad ecophysiological implications thereof. This study explored the putative association between autophagy and heat-tolerance using Drosophila melanogaster as a model. It was hypothesized that (i) heat-stress would cause an increase of autophagy in flies' tissues, and (ii) rapamycin exposure would trigger a detectable autophagic response in adults and increase their heat-tolerance. In line with this hypothesis, flies exposed to heat-stress present signs of protein aggregation and appear to trigger an autophagy-related homoeostatic response as a result. It was further shown that rapamycin feeding causes the systemic effect associated with target of rapamycin (TOR) inhibition, induces autophagy locally in the fly gut, and increases the heat-stress tolerance of individuals. These results argue in favour of a substantial contribution of autophagy to the heat-stress tolerance mechanisms of insects. |
Yamada, T., Yoshinari, Y., Tobo, M., Habara, O., Nishimura, T. (2023). Nacalpha protects the larval fat body from cell death by maintaining cellular proteostasis in Drosophila. Nat Commun, 14(1):5328 PubMed ID: 37658058
Summary: Protein homeostasis (proteostasis) is crucial for the maintenance of cellular homeostasis. Impairment of proteostasis activates proteotoxic and unfolded protein response pathways to resolve cellular stress or induce apoptosis in damaged cells. However, the responses of individual tissues to proteotoxic stress and evoking cell death program have not been extensively explored in vivo. This study shows that a reduction in Nascent polypeptide-associated complex protein alpha subunit (Nacα) specifically and progressively induces cell death in Drosophila fat body cells. Nacα mutants disrupt both ER integrity and the proteasomal degradation system, resulting in caspase activation through JNK and p53. Although forced activation of the JNK and p53 pathways was insufficient to induce cell death in the fat body, the reduction of Nacα sensitized fat body cells to intrinsic and environmental stresses. Reducing overall protein synthesis by mTor inhibition or Minute mutants alleviated the cell death phenotype in Nacα mutant fat body cells. This work revealed that Nacα is crucial for protecting the fat body from cell death by maintaining cellular proteostasis, thus demonstrating the coexistence of a unique vulnerability and cell death resistance in the fat body. |
Clemot, M., D'Alterio, C., Kwang, A. C., Jones, D. L. (2024). mTORC1 is required for differentiation of germline stem cells in the Drosophila melanogaster testis.PLoS One, 19(3):e0300337 PubMed ID: 38512882
Summary: Metabolism participates in the control of stem cell function and subsequent maintenance of tissue homeostasis. How this is achieved in the context of adult stem cell niches in coordination with other local and intrinsic signaling cues is not completely understood. The Target of Rapamycin (TOR) pathway is a master regulator of metabolism and plays essential roles in stem cell maintenance and differentiation. In the Drosophila male germline, mTORC1 is active in germline stem cells (GSCs) and early germ cells. Targeted RNAi-mediated downregulation of mTor in early germ cells causes a block and/or a delay in differentiation, resulting in an accumulation of germ cells with GSC-like features. These early germ cells also contain unusually large and dysfunctional autolysosomes. In addition, downregulation of mTor in adult male GSCs and early germ cells causes non-autonomous activation of mTORC1 in neighboring cyst cells, which correlates with a disruption in the coordination of germline and somatic differentiation. This study identifies a previously uncharacterized role of the TOR pathway in regulating male germline differentiation. |
Khan, M. R., Yin, X., Kang, S. U., Mitra, J., Wang, H., Ryu, T., Brahmachari, S., Karuppagounder, S. S., Kimura, Y., Jhaldiyal, A., Kim, H. H., Gu, H., Chen, R., Redding-Ochoa, J., Troncoso, J., Na, C. H., Ha, T., Dawson, V. L., Dawson, T. M. (2023). Enhanced mTORC1 signaling and protein synthesis in pathologic α-synuclein cellular and animal models of Parkinson's disease. Science translational medicine, 15(724):eadd0499 PubMed ID: 38019930
Summary: Pathologic α-synuclein plays an important role in the pathogenesis of &alpha-synucleinopathies such as Parkinson's disease (PD). Disruption of proteostasis is thought to be central to pathologic α-synuclein toxicity; however, the molecular mechanism of this deregulation is poorly understood. Complementary proteomic approaches in cellular and animal models of PD were used to identify and characterize the pathologic α-synuclein interactome. This study reports that the highest biological processes that interacted with pathologic α-synuclein in mice included RNA processing and translation initiation. Regulation of catabolic processes that include autophagy were also identified. Pathologic α-synuclein was found to bind with the tuberous sclerosis protein 2 (TSC2) and to trigger the activation of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1), which augmented mRNA translation and protein synthesis, leading to neurodegeneration. Genetic and pharmacologic inhibition of mTOR and protein synthesis rescued the dopamine neuron loss, behavioral deficits, and aberrant biochemical signaling in the α-synuclein preformed fibril mouse model and Drosophila transgenic models of pathologic α-synuclein-induced degeneration. Pathologic α-synuclein furthermore led to a destabilization of the TSC1-TSC2 complex, which plays an important role in mTORC1 activity. Constitutive overexpression of TSC2 rescued motor deficits and neuropathology in α-synuclein flies. Biochemical examination of PD postmortem brain tissues also suggested deregulated mTORC1 signaling. These findings establish a connection between mRNA translation deregulation and mTORC1 pathway activation that is induced by pathologic α-synuclein in cellular and animal models of PD. |
Liang, Y., Pan, C., Yin, T., Wang, L., Gao, X., Wang, E., Quang, H., Huang, D., Tan, L., Xiang, K., Wang, Y., Alexander, P. B., Li, Q. J., Yao, T. P., Zhang, Z., Wang, X. F. (2024). Branched-Chain Amino Acid Accumulation Fuels the Senescence-Associated Secretory Phenotype. Adv Sci (Weinh), 11(2):e2303489 PubMed ID: 37964763
Summary: The essential branched-chain amino acids (BCAAs) leucine, isoleucine, and valine play critical roles in protein synthesis and energy metabolism. Despite their widespread use as nutritional supplements, BCAAs' full effects on mammalian physiology remain uncertain due to the complexities of BCAA metabolic regulation. Here a novel mechanism linking intrinsic alterations in BCAA metabolism is identified to cellular senescence and the senescence-associated secretory phenotype (SASP), both of which contribute to organismal aging and inflammation-related diseases. Altered BCAA metabolism driving the SASP is mediated by robust activation of the BCAA transporters Solute Carrier Family 6 Members 14 and 15 as well as downregulation of the catabolic enzyme BCAA transaminase 1 during onset of cellular senescence, leading to highly elevated intracellular BCAA levels in senescent cells. This, in turn, activates the mammalian target of rapamycin complex 1 (mTORC1) to establish the full SASP program. Transgenic Drosophila models further indicate that orthologous BCAA regulators are involved in the induction of cellular senescence and age-related phenotypes in flies, suggesting evolutionary conservation of this metabolic pathway during aging. Finally, experimentally blocking BCAA accumulation attenuates the inflammatory response in a mouse senescence model, highlighting the therapeutic potential of modulating BCAA metabolism for the treatment of age-related and inflammatory diseases. |
Huang, J., Zhou, F., Zhou, H., Zheng, X., Huo, Z., Yang, M., Xu, Z., Liu, R., Wang, L., Wang, X. (2023). Systematic assessment of transcriptomic and metabolic reprogramming by blue light exposure coupled with aging. PNAS nexus, 2(12):pgad390 PubMed ID: 38059264
Summary: The prevalent use of light-emitting diodes (LEDs) has caused revolutionary changes in modern life, but the potential hazards to health of blue light are poorly understood. N(6)-methyladenosine (m(6)A) is the most prevalent posttranscriptional modification in eukaryotes and can modulate diverse physiological processes by regulating mRNA fate. To understand the effects and molecular mechanisms of daily low-intensity blue light exposure (BLE) and ascertain whether m(6)A methylation plays a role in BLE-induced phenotypes, this study constructed a series of Drosophila models under different durations of daily low-intensity BLE and multiomics profiles were obtained. These results revealed that BLE could induce transcriptomic, m(6)A epitranscriptomic, and metabolomic reprogramming in Drosophila along with aging process. Importantly, the m(6)A methylation sites enriched in the 5' untranslated regions (UTRs) of Drosophila transcripts showed strong age specificity and could be altered by BLE. It was experimentally validated that aging-related gene Tor and circadian rhythm-related gene per were regulated by 5' UTR-enriched m(6)A methylation. Overall, this study provides a systematic assessment of m(6)A RNA methylome reprogramming by BLE and aging in Drosophila model. |
Zhang, S., Wu, S., Yao, R., Wei, X., Ohlstein, B., Guo, Z. (2024). Eclosion muscles secrete ecdysteroids to initiate asymmetric intestinal stem cell division in Drosophila. Dev Cell, 59(1):125-140.e112 PubMed ID: 38096823
Summary: During organ development, tissue stem cells first expand via symmetric divisions and then switch to asymmetric divisions to minimize the time to obtain a mature tissue. In the Drosophila midgut, intestinal stem cells switch their divisions from symmetric to asymmetric at midpupal development to produce enteroendocrine cells. However, the signals that initiate this switch are unknown. This study identified the signal as ecdysteroids. In the presence of ecdysone, EcR and Usp promote the expression of E93 to suppress Br expression, resulting in asymmetric divisions. Surprisingly, the primary source of pupal ecdysone is not from the prothoracic gland but from dorsal internal oblique muscles (DIOMs), a group of transient skeletal muscles that are required for eclosion. Genetic analysis shows that DIOMs secrete ecdysteroids during mTOR-mediated muscle remodeling. These findings identify sequential endocrine and mechanical roles for skeletal muscle, which ensure the timely asymmetric divisions of intestinal stem cells. |
Lottes, E. N., Ciger, F., Bhattacharjee, S., Timmins, E. A., Tete, B., Tran, T., Matta, J., Patel, A. A., Cox, D. N. (2024). CCT and Cullin1 Regulate the TORC1 Pathway to Promote Dendritic Arborization in Health and Disease. Cells, 13(12) PubMed ID: 38920658
Summary: The development of cell-type-specific dendritic arbors is integral to the proper functioning of neurons within their circuit networks. This study examined the regulatory relationship between the cytosolic chaperonin CCT, key insulin pathway genes, and an E3 ubiquitin ligase (Cullin1) in dendritic development. CCT loss of function (LOF) results in dendritic hypotrophy in Drosophila Class IV (CIV) multi-dendritic larval sensory neurons, and CCT has recently been shown to fold components of the TOR (Target of Rapamycin) complex 1 (TORC1) in vitro. Through targeted genetic manipulations, this study confirm that an LOF of CCT and the TORC1 pathway reduces dendritic complexity, while overexpression of key TORC1 pathway genes increases the dendritic complexity in CIV neurons. Furthermore, both CCT and TORC1 LOF significantly reduce microtubule (MT) stability. CCT has been previously implicated in regulating proteinopathic aggregation, thus, this study examined CIV dendritic development in disease conditions as well. The expression of mutant Huntingtin leads to dendritic hypotrophy in a repeat-length-dependent manner, which can be rescued by Cullin1 LOF. Together, these data suggest that Cullin1 and CCT influence dendritic arborization through the regulation of TORC1 in both health and disease. |
Chen, H. Y., Wu, P. S., Li, Z. Y., Liu, Y. C., Yeh, S. R., Duan, B. C., Cheng, K. W., Hsu, C. C., Chiu, Y. L., Lee, W. T., Fan, S. Z., Wang, P. Y. (2025). Gut microbiome and host TOR pathway interact to regulate predator-induced aversive memory in Drosophila melanogaster. Proc Natl Acad Sci U S A, 122(25):e2422928122 PubMed ID: 40540603
Summary: The gut microbiome has emerged as a key factor influencing a wide range of host physiological processes and behaviors, though the mechanisms behind these effects remain only partially understood. This study explored the role of the gut microbiome in memory regulation using a parasitoid wasp-induced oviposition depression paradigm in Drosophila melanogaster. These findings show that flies with depleted gut microbiota, either through axenic culture or antibiotic treatment, exhibited significant memory impairments. However, reintroducing the commensal bacterium Lactobacillus plantarum alone was sufficient to restore memory, while coinoculation with Acetobacter pomorum further enhanced memory performance. Hemolymph metabolomic analyses revealed reduced amino acid levels in antibiotic-treated flies, which were linked to impaired Drosophila target of rapamycin (dTOR) signaling. Additionally, genetic manipulation of dTOR or dietary supplementation with branched-chain amino acids either mimicked or rescued the memory deficits caused by antibiotic treatments. These results suggest that the gut microbiome is essential for regulating memory function by maintaining amino acid homeostasis and proper dTOR signaling, with profound implications for advancing knowledge of cognitive regulation. |
Cao, Z., Zhang, C., Liu, L., Lei, H., Zhang, H., He, Y., Li, X., Xiang, Q., Wang, Y. F., Zhang, L., Chen, G. (2025). Microbiota-derived indole acetic acid extends lifespan through the AhR-Sirt2 pathway in Drosophila. mSystems, 10(5):e0166524 PubMed ID: 40197001
Summary: Disruption of aryl hydrocarbon receptor (AhR) signaling and aberrant tryptophan metabolism have been shown to be highly associated with aging and age-related disorders. However, the underlying molecular mechanisms by which the AhR-mediated signaling pathway contributes to the aging process remain largely unknown. This study finds that aged Drosophila exhibits markedly reduced tryptophan metabolism leading to impaired AhR ligands, especially indole acetic acid (IAA), compared with their young controls. Supplementation with IAA, produced from Lactobacillus spp., dose-dependently extends the lifespan of Drosophila and improves healthy aging with resistance to starvation and oxidative stress. Mechanistically, activation of AhR by IAA markedly enhances Sirt2 activity by binding to its promoter, thereby inhibiting downstream TOR signaling and related fatty acid and amino acid metabolism. Both Ahr and Sirt2 mutant flies with IAA supplementation display a negligible lifespan extension, suggesting that AhR-mediated Sirt2 signaling contributes to lifespan extension in flies upon IAA supplementation. From the perspective of host metabolism, IAA supplementation significantly increases unsaturated fatty acids (UFAs) in aged flies, which are regarded to be beneficial for healthy status. These findings provide new insights into the physiological functions of AhR involved in the aging process by mediating Sirt2 signaling. |
The adaptation of growth in response to nutritional changes is essential for the proper development of all organisms. Described here is the identification of the Drosophila homolog of the target of rapamycin (TOR, often referred to as dTOR), a candidate effector for nutritional sensing. Genetic and biochemical analyses indicate that TOR impinges on the insulin signaling pathway by autonomously affecting growth through modulating the activity of Drosophila S6k. However, in contrast to other components in the insulin signaling pathway, partial loss of TOR function preferentially reduces growth of the endoreplicating tissues. TOR is required cell autonomously for normal growth and proliferation during larval development, and for increases in cellular growth caused by activation of the phosphoinositide 3-kinase (PI3K) signaling pathway. The kinase activity of TOR is required for growth factor-dependent phosphorylation of S6 kinase. Loss of TOR results in cellular phenotypes characteristic of amino acid deprivation, including reduced nucleolar size, lipid vesicle aggregation in the larval fat body, and a cell type-specific pattern of cell cycle arrest that can be bypassed by overexpression of the S-phase regulator cyclin E. These results suggest that TOR regulates growth during animal development by coupling growth factor signaling to nutrient availability (Oldham, 2000b; Zhang, 2000).
During the development of unicellular and multicellular organisms, growth is dependent on the integration of diverse intracellular signals, which are triggered by patterning and/or environmental cues (Oldham, 2000a). The availability of nutrients strongly influences growth of single cells and multicellular organisms and in some cases specifies alternative developmental programs. For example, in yeast, nitrogen or carbon deprivation leads to withdrawal from vegetative growth, arrest in G0, and up-regulation of autophagy. Likewise, in the nematode Caenorhabditis elegans, limiting nutrients or overcrowding elicits an alternative developmental program termed the dauer stage. In this stage, the animal remains sexually immature and stockpiles additional lipids for survival during unfavorable growth periods. In more complex organisms such as insects and mammals, an alternative developmental program is lacking. Instead, nutrient limitation is managed by delaying development and, in severe cases, reducing the final body size of the organism. In the case of Drosophila, when nutrients become limiting, available resources are mobilized toward maintaining the growth of the mitotic tissues, which during metamorphosis form smaller, yet still fertile, flies (Oldham, 2000b and references therein).
In metazoans, one component of the complex physiological response of energy homeostasis and growth control is the insulin and IGF signaling system. Insulin and IGF activate two main signaling pathways via the insulin receptor substrates (IRS1-4): the Ras/MAPK pathway, which is involved in proliferation, and the phosphatidyl-inositol 3-kinase (PI3K) signaling pathway, which is involved in cell growth, survival, and metabolic homeostasis. PI3K mediates its effects on downstream signaling components through the production of phosphatidylinositol 3,4,5-tris phosphate (PIP3), which acts to recruit pleckstrin homology (PH) domain-containing proteins like protein kinase B (PKB). The actions of activated PI3K are antagonized by the 3'-phospho-inositol specific lipid phosphatase encoded by the tumor suppressor gene PTEN. An important protein implicated in the PI3K signaling pathway is the target of rapamycin (TOR). TOR has been reported to be regulated by PKB (Scott, 1998). Initially, TOR1 and TOR2 were identified in yeast as mutations that conferred resistance to the antiproliferative effects of rapamycin (Heitman, 1991). Rapamycin is an antibiotic that inhibits both yeast TOR and mammalian TOR (mTOR, also known as FRAP or RAFT) function by forming an inhibitory complex with the immunophilin, FK506 binding protein-12 (FKBP12), that binds to a region adjacent to the kinase domain termed the FKBP12-rapamycin binding domain (FRB; Brown, 1994; Sabatini, 1994; Thomas, 1997; Cutler, 1999). TOR is most related to the ATM/DNA-PK family of checkpoint protein kinases and is more distantly related to the PI3K family (Thomas, 1997; Cutler, 1999; Oldham, 2000b and references therein).
A key downstream target of mTOR function is protein synthesis. In part, mTOR positively mediates protein synthesis by modulating the activities of important translational components, including the translation initiation factor 4E binding proteins (4E-BP1-3) and the ribosomal protein S6 kinases (Chou, 1995; Lawrence, 1997; Dennis, 1999). Under conditions of reduced nutrients, such as amino acid limitation, mTOR negatively regulates protein synthesis and positively up-regulates autophagy (Dennis, 1999). Thus, mTOR may serve as a nutritional checkpoint for cell growth and ultimately, proliferation (Oldham, 2000b and references therein).
To initiate a genetic analysis of TOR in a multicellular organism, mutations were generated in Drosophila TOR. These mutants were used to study the role of TOR during development. The PI3K/Akt/p70S6K signaling module is conserved in Drosophila, where it acts to regulate cell, organ, and organismal growth (for review, see Coelho and Leevers 2000). Mutational inactivation of this pathway reduces cell size, hinders proliferation, and delays or arrests development, and its activation leads to autonomous increases in cell and organ size. Drosophila TOR mutant phenotypes are found to recapitulate aspects of both PI3K-dependent signaling and nutritional sensing, consistent with TOR acting at the junction of these pathways (Oldham, 2000b; Zhang, 2000).
A tissue-specific genetic screen has been carried out for recessive mutations affecting cell growth and proliferation in the Drosophila compound eye. In this screen, genetically mosaic flies are generated in which the eye and head capsule are homozygous for a randomly induced mutation, while the rest of the body and the germ line are heterozygous and, thus, phenotypically wild type. Remarkably, mosaic flies containing a mutation in a growth-promoting gene have eye and head structures that are strongly reduced in size relative to their wild-type sized heterozygous bodies and are termed pinhead flies. Two EMS-induced pinhead mutations (2L1 and 2L19) map to chromosomal position 34A, where the Drosophila homolog of TOR (dTOR) is located. These mutations fail to complement two lethal P-element insertions, EP(2)2353 and l(2)k17004, located 262 and 211 bp, respectively, upstream of the putative translation start site of dTOR. Sequence analysis of DNA from flies heterozygous for the EMS-induced dTOR mutations reveal two nucleotide substitutions. The lesion of dTOR2L1 results in a change of a proline to a leucine at amino acid position 2303 (P2303L). The location of this mutation within a highly conserved region of the kinase domain implies that the kinase activity of dTOR is critical for its function, as has been shown for the yeast TORs. In contrast, the lesion in dTOR2L19 is an arginine changed to a nonsense mutation at amino acid residue 248 (R248Stop), giving rise to a stop codon. This mutation would be predicted to result in a short, truncated protein and should thus be a complete loss-of-function mutation (Oldham, 2000b).
The phenotypes associated with the complete loss of dTOR function are remarkably similar to phenotypes associated with mutations in the Insulin-like receptor (Inr) pathway. (1) Strong dTOR mutants arrest development at a similar stage as do strong mutants in the Inr pathway or amino acid-starved larvae with little detectable imaginal tissue. (2) dTOR mutant clones have a significant proliferative disadvantage similar to Inr pathway mutant clones. Clones of dTOR null mutant cells, although severely affected, are not cell lethal. Similarly, in most mammalian cell types, rapamycin decreases but does not abolish cell growth, except for IL2-mediated T-cell proliferation. (3) The strict autonomous control of cell growth without disturbing the specification and differentiation is also seen with the dTOR mutants. Indeed, loss of dTOR function in clones of homozygous mutant cells in the adult eye show that only the mutant cells, as exemplified by the dark, circular rhabdomeres, are severely reduced in size. Analysis of imaginal wing disc cells at the end of the third larval instar by fluorescence-activated cell sorting (FACS), confirms that cells from the weak heteroallelic combination, dTOR2L1/dTORl(2)k17004, are smaller than those of wild type. The effect on cell size is more pronounced in G1 than G2, consistent with mTOR function having a predominante role on cell growth during G1 (Oldham, 2000b).
Despite this observation, there is no apparent difference between the distribution of dTOR mutant and wild-type cells within each phase of the cell cycle. Although the similarities of the loss-of-function mutant phenotypes of dTOR and other components of the Inr pathway are consistent with a model in which dTOR acts downstream of dPI3K, the analysis of partial loss-of-function dTOR mutants and the biochemical analysis of Drosophila S6K activity indicates a more complex relationship between dTOR and the Inr pathway (Oldham, 2000b).
Genetic interactions between dTOR mutants and other mutations in the insulin pathway were examined. Drosophila Pten encodes a negative effector of insulin signaling, and eyes and heads lacking Pten function are significantly larger than wild-type heads. Removal of dTOR function strongly reduces the size of Pten mutant heads, suggesting that dTOR is required for the increased growth generated by the loss of Pten function (Oldham, 2000b).
In vertebrates, S6K activity is blocked by rapamycin, an inhibitor of TOR. Therefore, Drosophila S6K activity was examined in immunoprecipitates of extracts from larvae mutant for dTOR, S6K, chico, and larvae treated with rapamycin or deprived of amino acids. A severe reduction in the phosphorylation of ribosomal protein S6 was observed in extracts from strong dTOR2L1/dTOR2L19 mutant larvae. This was not caused by a reduction in Drosophila S6k protein as shown by Western blotting of these extracts. In addition, the S6k protein is up-regulated in the dTOR mutant larvae and amino acid-starved larvae. In all cases, Western blot analysis has shown equivalent amounts of initiation factor 4E (eIF-4E). S6k activity is not detected in S6kl-1 null mutants and is severely reduced when wild-type larvae are starved for amino acids or treated with rapamycin. Higher doses of rapamycin blocks development during early larval stages, leading to lethality. Analysis of the weak dTOR2L1/dTORl(2)k17004 or dTOR2L1/dTOREP(2)2353 heteroallelic combinations also reveal a reduction in S6k activity in the third larval instar and an up-regulation of the protein as compared with wild-type flies. The surprising fact that dTOR mutants and amino acid starvation result in an up-regulation of S6k levels suggests that dTOR and amino acids may negatively control the protein levels of S6k. Unexpectedly, S6k activity as well as protein levels are unaffected in chico mutants. It may be that Inr does not signal to S6k or that S6k resides on a parallel pathway that bifurcates upstream of Chico. In support of the latter possibility, Inr has been shown to genetically interact with PI3K independently of Chico, presumably through docking sites for the p60 adaptor of PI3K in the Inr C-terminal tail. This result suggests that there is a S6k independent pathway for growth control and that the reduced Inr-mediated PI3K signaling in a chico mutant is sufficient for S6k activation (Oldham, 2000b).
The biochemical differences between the ability of Chico and dTOR to activate S6k argue for a more complex relationship between the Inr pathway and dTOR. Given the low number of pharate adults, the weights of dTOR, S6kl-1, and chico mutants were compared at an early pupal stage. The weight of the dTOR mutant pupae is more similar to S6k than to chico mutant pupae. Thus, in the absence of S6k function or the presence of reduced dTOR levels, cellular growth rates are diminished but larvae pupariate at a larger size as a result of a longer developmental delay. Importantly, S6k mutant flies have cells that are smaller but of the normal number. However, in chico mutants, pupariation is initiated at a much smaller size. The result is that chico mutants emerge after only a 2-d delay and are smaller than dTOR and S6k mutants because of fewer and smaller cells. Therefore, while insulin signaling controls cell size and cell number, S6k primarily controls cell size. It will be of interest to know whether dTOR is also limited to controlling only cell size (Oldham, 2000b).
Larvae are composed of mitotic cells, largely represented by the imaginal discs, and of endoreplicating tissues, which form larval structures like the gut, fat body, and salivary glands. An increase in DNA ploidy of larval cells is required for the ~200-fold increase in mass obtained by the larvae during the 5-d period between the completion of embryogenesis and the beginning of pupation. During starvation, larvae sacrifice their endoreplicating tissue to maintain the growth and proliferation of the mitotic cells that are required to form the reproductive adult. Furthermore, S6k activity is reduced in starved larvae and dTOR mutants. These observations prompted an analysis of the mitotic and endoreplicating tissues of dTOR, S6k, and chico mutant larvae just before pupariation. Strong dTOR and PI3K mutants, as well as amino acid-starved larvae, are incapable of growth and have barely detectable imaginal and endoreplicative tissues. Surprisingly, the wing discs of the weak dTOR heteroallelic combination are of approximately equivalent size to that of wild-type larvae, whereas those of S6kl-1 mutants are reduced. However, the amount of endoreplicating tissue in the dTOR mutant as compared to wild-type larvae is severely decreased. This is clearly demonstrated by comparing the salivary glands of dTOR mutant and wild-type larvae. In contrast, the size of endoreplicating tissue and imaginal discs in S6k null mutants as well as chico null mutants is reduced to approximately the same extent. Staining of the salivary glands with DAPI and phalloidin reveals that the size of the nuclei and, thus, the degree of endoreplication is severely reduced in S6k, chico, and dTOR mutants. The difference in size between dTOR and S6k mutant salivary glands is largely caused by a very pronounced reduction in cytoplasmic volume in dTOR mutants. The nuclear to cytoplasmic ratio is higher in dTOR salivary glands than in y w, S6k, or chico mutant salivary glands. Thus, it appears that partial loss of dTOR function permits the growth of imaginal tissue to wild-type size, while endoreplicating tissue is disproportionally reduced, a phenotype distinct from S6k mutants. Consistent with this finding, the lethality of the different dTOR mutants could not be rescued by constitutive expression of a S6K1 variant, D3E-E389, which exhibits high basal activity in the absence of mitogens under the control of the alpha-tubulin promoter, which rescues all aspects of the S6kl-1 null phenotype. Therefore, S6k-independent processes must contribute to the weak dTOR phenotype (Oldham, 2000b).
The effect of rapamycin and amino acids on translation in mammals is mediated through the S6Ks and the 4E-BPs. Unlike the other elements in the PI3K signaling pathway, absence of amino acids blocks both S6K activation and 4E-BP phosphorylation. Indeed, a mutant of S6K1, lacking a portion of both its amino and carboxyl termini, is resistant to rapamycin but still sensitive to the fungal metabolite wortmannin, an inhibitor of PI3K. This suggests that the PI3K-dependent signal to S6K activation does not involve TOR. This same mutant is also unaffected by amino acid withdrawal, consistent with the role of mTOR as an amino acid checkpoint in S6K activation. Although there is some controversy concerning the ability of mitogens to activate mTOR, the in vitro activity of mTOR from cultured cells toward either itself, S6K1, or 4E-BP1 is unaffected by mitogens. Thus, mTOR may act as a permissive signal that primes 4E-BP phosphorylation and S6K activation by the PI3K signaling pathway if amino acids, and possibly other nutrients, are at sufficient levels. Likewise, in Drosophila larvae, amino acids are necessary, but not sufficient, for imaginal disc and endoreplicating tissue proliferation, compatible with dTOR acting in a parallel pathway involved in amino acid sensing. The fact that chico mutant larvae have normal levels of S6k activity and that the dTOR larval phenotypes with respect to the imaginal discs and endoreplicating tissues are so distinct compared with other mutants in the Inr pathway, supports the possibility that dTOR is not responsive to insulin signaling (Oldham, 2000b and references therein).
It is well established in yeast that TOR is an important mediator of nutrient limitation, and it has been proposed that TOR acts as an amino acid effector to coordinate the response of yeast to different nutritional conditions (Barbet, 1996). Indeed, the similarities between dTOR mutant larvae and larvae deprived of amino acids are striking. Therefore, it is likely that dTOR also functions as an amino acid sensor in multicellular organisms. The fact that yeast and Arabidopsis do not have an insulin system suggests that TOR may be an ancestral and widespread nutritional sensor. To provide additional levels of control, it may have been integrated into the insulin system later to respond to different modes of nutrient deprivation with different developmental responses (Oldham, 2000b and references therein).
Insulin treatment of Drosophila Kc 167 cells induces the multiple phosphorylation of a Drosophila ribosomal
protein, as judged by its decreased electrophoretic mobility on two-dimensional polyacrylamide gels. The extent to which insulin
induces this response is potentiated by cycloheximide and blocked by pretreatment with rapamycin. Isolation and mass
spectrometric analysis have revealed that the multiply phosphorylated protein is the larger of two Drosophila melanogaster
orthologs of mammalian 40S ribosomal protein S6, termed here DS6A. Proteolytic cleavage of DS6A (derived from
stimulated Kc 167 cells), with the endoproteinase Lys-C releases a number of peptides, one of which contains all the putative
phosphorylation sites. Conversion of phosphoserines to dehydroalanines with Ba(OH)(2) shows that the sites of
phosphorylation reside at the carboxy terminus of DS6A. The sites of phosphorylation have been identified by Edman degradation
after conversion of the phosphoserine residues to S-ethylcysteine as Ser(233), Ser(235), Ser(239), Ser(242), and Ser(245).
Phosphopeptide mapping of individual phosphoderivatives, isolated from two-dimensional polyacrylamide gels,
indicate that DS6A phosphorylation, in analogy to mammalian S6 phosphorylation, appears to proceed in an ordered fashion (Oldham, 2000b and references therein).
The evolutionarily conserved serine/threonine protein kinase target-of-rapamycin (TOR) controls cell growth as a core component of TOR complexes 1 (TORC1) and 2 (TORC2). Although TORC1 is the more central growth regulator, TORC2 has also been shown to affect cell growth. This study demonstrates that Drosophila LST8, the only conserved TOR-binding protein present in both TORC1 and TORC2, functions exclusively in TORC2 and is not required for TORC1 activity. In mutants lacking LST8, expression of TOR and RAPTOR, together with their upstream activator Rheb, was sufficient to provide TORC1 activity and stimulate cell and organ growth. Furthermore, using an lst8 knockout mutation, this study showed that TORC2 regulates cell growth cell autonomously. Surprisingly, however, TORC2 does not regulate cell growth via its best-characterized target, AKT. These findings support the possible application of TORC2-specific drugs in cancer therapy (Wang, 2012).
LST8 was originally identified genetically as a mutation in yeast that is synthetically lethal with sec13-1, a mutation that causes a sorting defect in the secretory pathway. Later, physical association studies showed that LST8 is a core component of TORC1 and TORC2 in both yeast and mammals. Structural data confirmed that LST8 stably interacts with the kinase domain of TOR without overlapping the Raptor binding site. While it has been clearly demonstrated that LST8 is essential for TORC2 activity, whether or not LST8 functioned in the TORC1 pathway had not been rigorously investigated. In mammalian cells, it was originally reported that the binding of LST8 to mTOR strongly stimulated the binding association between mTOR and Raptor and, as a result, elevated the kinase activity of mTORC1 toward S6K1 and 4E-BP1. In yeast, rapamycin, which is TORC1 specific, mimicked the Gap1p sorting defect of an lst8 mutant, and a temperature-sensitive lst8 mutant was hypersensitive to rapamycin, suggesting that LST8 functions in TORC1. However, the observations that the lst8 knockout did not affect the phosphorylation of S6K1 or 4E-BP1 and that lst8 knockout mice phenocopied rictor knockout mice but not mtor knockout mice suggested that LST8 might not function in TORC1, at least under normal conditions (Wang, 2012).
This report demonstrates that lst8 knockout flies are viable but small, similar to rictor mutants but dissimilar to files with tor or rheb mutations, which are lethal. Neither loss nor overexpression of LST8 affected the kinase activity of TORC1 toward S6K or autophagy, whereas the kinase activity of TORC2 toward AKT was completely lost in the lst8 mutants. Moreover, in the absence of LST8 the overexpression of Rheb still upregulated TORC1 activity and also promoted the growth of cells. Furthermore, the expression of TOR and Raptor was sufficient to drive phosphorylation of S6K and growth of cells when they were supplied with Rheb signaling, even when LST8 was entirely absent. Given these results, it is concluded that LST8 is an essential core component of TORC2 but is dispensable for the regulation and activity of TORC1 (Wang, 2012 and references therein).
Although TOR signaling functions primarily as a central regulator of cell growth, increasing evidence suggests that inappropriate activation of TOR also makes cells sensitive to death signals. For instance, tumor cells in TSC disease, which is caused by mutation of tsc1 or tsc2, exhibit morphological features of apoptosis such as activated caspase-8 and positive terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) results. In a mouse model of TSC disease, conditional knockout of tsc1 induced cell death in the hippocampus and neocortex. In a recent study in flies, it has been shown that activation of TORC1 by either overexpression of Rheb or mutation of Tsc1 led to neurodegeneration. In the latter case the neurodegeneration was specifically attributed to TOR's ability to suppress autophagy (Wang, 2012).
Although overactivation of TORC1 can result in cell death, it is not always sufficient for the induction of cell death. For instance, in young flies, overexpression of Rheb alone promoted cell growth but did not drive cell death, whereas neuronal cell death was detected only in aged flies after long-term TOR hyperactivation. This slow neurodegeneration was also dependent on neuronal activity. The present study, however, found that when Rheb and TORC1 are overexpressed together (a condition which very strongly increases TORC1 activity), severe cell death rapidly occurred in all cell types, even in young animals, without any dependency on neuronal activity. Therefore, it is suggested that the regulation of TORC1 activity is crucial for the survival of all cell types and that the incidence of cell death within a tissue may be correlated with TORC1 activity levels. The cell death-promoting function of TORC1 might explain why mutations of tsc1 or tsc2 lead to benign, rather than malignant, tumors (Wang, 2012).
In both mammals and invertebrates, it has been demonstrated that TORC2 regulates growth and metabolism. However, whether TORC2 affects organismal growth cell autonomously or indirectly via systemic effects had not been thoroughly evaluated. In C. elegans, rictor mutants have been shown to be developmentally delayed, small in body size, and short-lived. Targeted expression of Rictor in the intestines of these mutants could rescue these growth defects, and therefore it was suggested that TORC2 might act directly in the intestine to regulate cell mass and growth of the whole animal, non-cell autonomously. This study presents evidence supporting a cell-autonomous role for TORC2 in the growth of several different cell types. In genetic mosaics, cells homozygous for the lst81 mutation were smaller than adjacent wild-type control cells in the retina, wing epithelium, and fat body. Moreover, expression of LST8 in posterior wing compartments specially rescued growth of the posterior cells without affecting the anterior (Wang, 2012).
TORC1 consists of TOR, Raptor, LST8, and PRAS40 (proline-rich AKT substrate, 40 kDa), a negative regulator. mTORC1 purified with PRAS40 at substoichiometric levels has been shown to have kinase activity toward S6K1 and 4E-BP1 in vitro. Raptor-free mTORC1 appears unable to support 4E-BP1 phosphorylation but might be capable of phosphorylating full-length S6K1. Interestingly, the phosphorylation of S6K1 by Raptor-free mTORC1 could still be inhibited by FKBP12-rapamycin. Nevertheless, overexpression of TOR and Raptor together has been shown to significantly increase in vitro TORC1 kinase activity compared to overexpressed TOR alone. Overexpressed TOR has been found in some cancer patients, but the significance of this relative to disease progression or TORC1 activity levels has not been assessed. By overexpression of TORC1 components in flies, this study demonstrated that expression of all three positive components of TORC1 (TOR, Raptor, and LST8) cannot drive increased TORC1 activity or cell overgrowth. However, in the presence of the upstream activator, Rheb, overexpression of TOR and Raptor either with or without LST8 increased TORC1 activity toward S6K and dramatically stimulated cell growth. These results further confirm that TOR and Raptor but not LST8 are bone fide components of TORC1. In contrast to results from in vitro studies, expression of TORC1 components did not function in the absence of activating signals, perhaps because cellular translocation of TORC1 is a key event in TORC1 activation. Since expression of TOR alone did not generate functional TORC1, the significance of overexpression of TOR in cancer cells might be reconsidered (Wang, 2012).
The fact that the two TOR complexes display distinct cellular functions and phosphorylate different downstream substrates suggests that they might respond to different upstream signals. It has been well established that TORC1 can be regulated by upstream signals such as amino acids and growth factors, but little is known about whether TORC2 can be activated by such signals. When purified from insulin- or IGF1-stimulated cells and assayed in vitro, TORC2 kinase activity is elevated, suggesting that growth factors can activate TORC2 through PI3K signaling. Data from tissue culture cells suggest that the TSC complex physically associates with TORC2 via Rictor, but surprisingly the regulation of TORC2 by the TSC1/2 complex appeared to be independent of its GTPase-activating protein activity toward Rheb. This study shows that AKT phosphorylation is downregulated when Rheb is overexpressed, suggesting a role for the Rheb GTPase in regulating TORC2. The recent finding that Rictor is directly phosphorylated by the mTORC1-dependent kinase S6K1 might help explain the regulation of TSC2 by Rheb. However, it is also possible that through negative feedback from TORC1, which phosphorylates and deactivates insulin receptor substrate 1, TORC2 is negatively regulated by Rheb signaling (Wang, 2012).
Nutrient availability is one of the major environmental signals influencing growth. Conflicting data have been proffered as to whether amino acids might regulate TORC2 activity. Although amino acids originally were not considered regulation signals for TORC2, it was recently reported that amino acids could increase AKT phosphorylation by TORC2. In addition, leucine addition to starved cells promotes cell migration in a TORC2-dependent manner. The fat body is an insect organ that retains the endocrine and storage functions of the vertebrate liver and adipose tissues and serves as a nutrient sensor that restricts global growth. Thus, fat body cells are very sensitive to nutritional conditions, and since TORC1 is controlled by nutrition, its activity has a profound influence in fat body cells. For instance, downregulation of TORC1 by expression of TSC1/2 strongly decreases fat body cell size relative to other tissues, where it has modest effects. In addition, fat body cells with upregulated TORC1 are capable of massive growth under starvation conditions, essentially bypassing the normal cessation of growth caused by starvation, whereas deregulated TORC1 has mild effects in fat body cells in fed animals. As this study shows, the disruption of TORC2 by mutation of lst8 affected the growth of every tissue to a similar extent. Moreover, starvation did not change the size reduction in lst81 cells in the fat body, suggesting that TORC2-regulated cell growth might not be nutrition sensitive (Wang, 2012).
TORC2 is believed to control cell survival, cell growth, and cytoskeletal organization by phosphorylating several AGC kinases including SGK, cPKCα, and AKT. Among these TORC2 substrates, AKT is especially important because of its general role in growth and survival. Therefore, it has been proposed that TORC2 might serve as a drug target in tumors that have lost the expression of PTEN, a tumor suppressor that inhibits AKT activity. However, recent evidence suggests that TORC2-mediated phosphorylation of AKT does not determine its absolute activity but, instead, determines substrate specificity. AKT variants immune to TORC2-mediated phosphorylation retain the ability to control glycogen synthase kinase 3 (GSK3) and TSC2 but show decreased activity toward FOXO1/3a. These observations align with those of this study in supporting the view that TORC2 might only help to inactivate the FOXO branch of the AKT signaling pathway without affecting the TORC1 branch (Wang, 2012).
The results of this study suggest that TORC2-mediated AKT phosphorylation does not regulate the growth of cells and argue against the importance of AKT as an important downstream target of TORC2 in cell growth control. Expression of nonphosphorylatable mutant forms of AKT did not change cellular growth status either with or without TORC2, and a constitutively active phospho-mimetic form of AKT was unable to suppress the growth defect of lst8 mutants. Moreover, mutation of foxo did not compensate for the reduction in cell growth caused by disruption of TORC2. In fact, similar results have been reported in C. elegans, where the reduced size of rictor mutants was proposed not to be a consequence of reduced AKT activity. Meanwhile, it was suggested that TORC2 controls cell growth by affecting SGK activity. However, the lack of an SGK homolog in flies casts some doubt on this mechanism. The unexpected finding of this study that AKT does not mediate TORC2-regulated cell growth emphasizes the importance of finding TORC2's critical substrates and also raises questions about the efficacy of TORC2 inhibitors as drugs for cancer therapy (Wang, 2012).
TORC1, in Drosophila consisting of Target of rapamycin, Raptor and Lst8) regulates growth and metabolism, in part, by influencing transcriptional programs. This study has identified REPTOR and REPTOR-BP, both leucine zipper DNA-binding proteins, as transcription factors downstream of TORC1 that are required for approximately 90% of the transcriptional induction that occurs upon TORC1 inhibition in Drosophila. Thus, REPTOR and REPTOR-BP are major effectors of the transcriptional stress response induced upon TORC1 inhibition, analogous to the role of FOXO downstream of Akt. When TORC1 is active, it phosphorylates REPTOR on Ser527 and Ser530, leading to REPTOR cytoplasmic retention. Upon TORC1 inhibition, REPTOR becomes dephosphorylated in a PP2A-dependent manner, shuttles into the nucleus, joins its partner REPTOR-BP to bind target genes, and activates their transcription. In vivo functional analysis using knockout flies reveals that REPTOR and REPTOR-BP play critical roles in maintaining energy homeostasis and promoting animal survival upon nutrient restriction (Tiebe, 2015).
Target of rapamycin complex 1 (TORC1) integrates information on energy and nutrient status in eukaryotic cells. Under high-nutrient and -energy conditions, TORC1 drives translation, ribosome biogenesis, mitochondrial activity, lipid synthesis, nucleotide synthesis, and glycolysis. TORC1, thereby, couples activity of cellular anabolic and catabolic pathways to nutrient and energy supply. TORC1 is frequently mis-regulated in diseases such as cancer, diabetes, obesity, and neurodegeneration (Tiebe, 2015).
TORC1 regulates growth and metabolism by phosphorylating target proteins, such as S6K and 4E-BP, involved in translational regulation. Phosphorylation of targets changes very rapidly upon altered TORC1 activity, allowing cells to adapt quickly to changing environmental conditions. In addition, TORC1 also has long-lasting impact on cellular behavior through the control of transcriptional programs. This occurs by directly or indirectly modulating the activity of transcription factors such as SREBP, HIF1a, PGC-1a, TIF1a, PPARa, Atf4 (CREB2), TFEB, and TFE3 (Tiebe, 2015).
The TORC1 signaling pathway is highly conserved through evolution, thereby enabling the use of model organisms such as Drosophila for discovery of novel pathway components. Recent studies in Drosophila analyzed the impact of TORC1 signaling on cellular transcription. In Drosophila S2 cells, inhibition of TORC1 with rapamycin leads to numerous transcriptional changes. Genes involved in anabolic processes such as ribosome biogenesis are strongly repressed upon TORC1 inhibition. Previous work showed that this occurs via downregulation of myc activity. A second class of genes is activated upon TORC1 inhibition. Although the function of these genes is less understood, they probably represent the genes needed for cells to adapt to conditions yielding reduced TORC1 activity, such as low nutrient availability. This study aimed to find the transcription factor responsible for mediating this upregulation upon TORC1 inhibition. The discovery is reported of these factors, which, surprisingly, are required for mediating most of the transcriptional induction that takes place upon TORC1 inhibition and play important roles in maintaining energy homeostasis in vivo (Tiebe, 2015).
This study has identify two uncharacterized genes, CG13624 and CG18619, which are termed REPTOR and REPTOR-BP, respectively, as transcription factors mediating circa 90% of the transcriptional repression downstream of TORC1 in Drosophila, which indicates that they are major effectors of TORC1. REPTOR is inhibited by TORC1-mediated phosphorylation and cytoplasmic retention by 14-3-3 proteins. Upon nutrient deprivation and low TORC1 activity, REPTOR becomes active, accumulating in the nucleus and binding target genes, a process that requires its partner, REPTOR-BP (Tiebe, 2015).
REPTOR is repressed when TORC1 activity is high, as is the case during larval stages when animals are feeding and growing. Hence, genetic removal of REPTOR during larval stages of well-fed animals has little phenotypic consequences, including no growth defects. In contrast, REPTOR is somewhat activated in (1) pupae and adults, which eat significantly less than larvae; (2) in larvae growing in low-nutrient conditions; and (3) in S2 cells growing in standard culture conditions. Hence, under these conditions, REPTOR loss of function leads to transcriptional and physiological phenotypes. The strongest REPTOR phenotypes become apparent when animals are starved, as these are the conditions where TORC1 is most inactive and, hence, REPTOR is most active (Tiebe, 2015).
The REPTOR regulatory system is analogous to another nutrient sensitive pathway—that of Akt and FOXO. When nutrients are low, Akt becomes inactive due to reduced systemic insulin signaling. This leads to FOXO dephosphorylation, release from 14-3-3 proteins, and nuclear accumulation, thereby activating target genes that mount a stress response. FOXO and REPTOR can be thought of as the respective counterparts for insulin and TORC1 signaling, which sense nutrients at the systemic and cell-autonomous levels, respectively. FOXO and REPTOR also have common target genes and bind near each other in shared enhancers. In sum, FOXO and REPTOR appear to have overlapping functions; indeed, genetic removal of both REPTOR and FOXO is synthetic lethal, indicating that they compensate for loss of each other (Tiebe, 2015).
As an effector of TORC1 function, REPTOR mediates some of the physiological effects of reduced TORC1 activity. Body-wide inhibition of TORC1 signaling leads to increased TAG levels and animals of reduced size. These phenotypes are, in part, due to activation of REPTOR, since removal of REPTOR strongly rescues them. Thus, TORC1 promotes growth during development not only by activating S6K but also by keeping REPTOR/REPTOR-BP repressed. Inhibition of TORC1 also extends lifespan. This could potentially be mediated, in part, via activation of REPTOR/REPTOR-BP, since both REPTOR and REPTOR-BP KO animals have significantly reduced lifespans (Tiebe, 2015).
Triglyceride levels were quantified in TOR2L1/2L19 hypomorphic larvae and found to be significantly elevated compared to those in controls, in line with a number of reports from adult flies. These results do not fit with one report that dTOR7/P mutant larvae are lean. The reason for this discrepancy or whether it has to do with the particular nature of the dTOR[7] and/or dTOR[P] alleles is unknown. Further work will be required to examine this carefully (Tiebe, 2015).
Both REPTOR and REPTOR-BP proteins have DBDs. Hence, the DNA-binding specificity of the REPTOR/REPTOR-BP dimer likely reflects the combined action of the two proteins. Since REPTOR-BP can also homodimerize, the REPTOR-BP homodimer might bind DNA with a pattern distinct from that of the REPTOR/REPTOR-BP dimer (Tiebe, 2015).
Many of the genes repressed by rapamycin in larvae (~80%) were no longer repressed by rapamycin in REPTOR KO larvae, raising the possibility that these genes are also REPTOR targets. It is not thought, however, that this is the case for several reasons. (1) In S2 cells, almost no genes require REPTOR to be repressed by rapamycin. If REPTOR were also involved in transcriptional repression, this would likely be seen also in S2 cells. (2) The REPTOR-induced genes are in common between S2 cells and larvae, whereas the REPTOR-repressed ones are not, suggesting their regulation might result from indirect effects. (3) Transactivation assays with REPTOR and REPTOR-BP only show strong transcriptional activation of the reporters but no repression. That said, many transcription complexes have both activating and repressive activities, so further investigation might find that REPTOR and REPTOR-BP also have repressive functions (Tiebe, 2015).
Surprisingly, REPTOR and REPTOR-BP have attracted little attention to date.
Microarray studies found that expression of CG13624 and CG18619 are regulated by nutrient conditions in the fly; however, no information regarding their function was available. Using BLAST to compare the human proteome with REPTOR and REPTOR-BP identifies Crebrf and Crebl2, respectively, which could potentially be human orthologs. It will be interesting to study them in light of the Drosophila data (Tiebe, 2015).
In summary, this study identifies REPTOR and REPTOR-BP as dedicated transcription factors that control the transcriptional repression downstream of TORC1 in Drosophila. Since these transcription factors mediate part of the functional output of TORC1, it will be interesting to assess their contribution toward the role that TORC1 plays in cancer, diabetes, and aging (Tiebe, 2015).
While beneficial effects of fasting on organismal function and health are well appreciated, little is known about the molecular details of how fasting influences synaptic function and plasticity. Genetic and electrophysiological experiments demonstrate that acute fasting blocks retrograde synaptic enhancement that is normally triggered as a result of reduction in postsynaptic receptor function at the Drosophila larval neuromuscular junction (NMJ). This negative regulation critically depends on transcriptional enhancement of eukaryotic initiation factor 4E binding protein (4E-BP) under the control of the transcription factor Forkhead box O (Foxo). Furthermore, these findings indicate that postsynaptic 4E-BP exerts a constitutive negative input, which is counteracted by a positive regulatory input from the Target of Rapamycin (TOR). This combinatorial retrograde signaling plays a key role in regulating synaptic strength. These results provide a mechanistic insight into how cellular stress and nutritional scarcity could acutely influence synaptic homeostasis and functional stability in neural circuits (Kauwe, 2016).
Many forms of dietary restriction can reduce cellular stress, improve organismal health, and in many instances extend lifespan in a number of model organisms. A major cellular function that is highly sensitive to nutrient intake from yeast to mammals is cap-dependent translation under the regulation of the target of rapamycin (TOR). TOR promotes cap-dependent translation primarily through phosphorylation of 4E-BPs (eukaryotic initiation factor 4E binding proteins) and p70 S6Ks (S6 ribosomal protein kinases). Phosphorylation of 4E-BP suppresses its ability to bind and inhibit the interaction between eIF4E (eukaryotic initiation factor 4E) and the initiation factor eIF4G, a critical step for translation initiation. In addition to the regulation by TOR, 4E-BP undergoes upregulation in response to dietary restriction and starvation. Together these two responses result in a strong inhibition of protein synthesis and act as a metabolic brake. Multiple lines of evidence suggest that fasting-induced increase in ketone bodies influences neuronal excitability and aspects of neurotransmitter release; however, little is known about how different forms of dietary restriction, by influencing protein translation, can exert an effect on the regulation of synaptic function and plasticity (Kauwe, 2016).
At the Drosophila larval neuromuscular junction (NMJ), the genetic removal of GluRIIA, one of five glutamate receptor subunits, reduces the postsynaptic response to unitary release of neurotransmitter. As a result of this reduced response to neurotransmitter, a retrograde signal is triggered in the postsynaptic muscle that ultimately leads to a compensatory enhancement in presynaptic release from the motor neuron, a process that is conserved at the vertebrate NMJs. The maintenance of this homeostatic synaptic compensation or retrograde synaptic enhancement is highly sensitive to postsynaptic cap-dependent translation in Drosophila; mutations in either Target of Rapamycin (TOR) or eIF4E can dominantly suppress the synaptic compensation in GluRIIA mutants (Penney, 2012). Interestingly, postsynaptic overexpression of TOR or S6K, in an otherwise wild-type muscle, is also sufficient to trigger a retrograde enhancement in presynaptic neurotransmitter release, suggesting that normal synaptic strength may be affected by a postsynaptic signal from the muscle (Penney, 2012; Kauwe, 2016 and references therein).
Previous findings have demonstrated that postsynaptic translation plays a critical role in the regulation of retrograde synaptic enhancement at the NMJ. Therefore, in light of the effect of dietary restriction on TOR-dependent translation, this study set out to investigate the consequence of nutrient restriction on retrograde synaptic compensation in GluRIIA mutants. Electrophysiological analysis indicates that acute fasting, but not amino acid restriction, blocks this retrograde synaptic compensation. This block is not merely due to reduced TOR activity, but rather a result of transcriptional upregulation of postsynaptic 4E-BP under the control of the transcription factor Foxo. These results indicate that the retrograde regulation of synaptic strength at the NMJ depends on the balance between 4E-BP and TOR (Kauwe, 2016).
A few hours of fasting can have a strong impact on retrograde synaptic enhancement at the Drosophila larval NMJ. Removal of food source acutely activates 4E-BP transcription in postsynaptic muscles in a Foxo-dependent manner, thereby leading to the inhibition of retrograde synaptic enhancement at the NMJ. The results indicate that Foxo and 4E-BP act cell autonomously in postsynaptic muscles to exert a retrograde negative regulation on presynaptic neurotransmitter release. Future studies are needed to test whether fasting-induced alterations in insulin signaling underlie the transcriptional upregulation of 4E-BP via its effect on Foxo in postsynaptic muscles.
While 4E-BP-mediated suppression of synaptic enhancement as a result of fasting could be considered undesirable during development, it can be beneficial under conditions of abnormally high synaptic activity. As such, 4E-BP-mediated inhibition of retrograde synaptic enhancement and the subsequent dampening of circuit activity might provide an explanation for the beneficial effects of fasting in reducing seizures in some cases. Similarly, in cases where dysregulation of TOR activity is thought to underlie abnormal circuit activity, such as in TSC models, intermittent fasting could potentially dampen the increase in synaptic release through a 4E-BP-dependent inhibition, thereby stabilizing neuronal circuits (Kauwe, 2016).
In addition to its role as a molecular responder to stress, 4E-BP exerts a constitutive negative regulation on presynaptic neurotransmitter release at the NMJ. Electrophysiological analysis of loss-of-function mutant larvae indicates that 4E-BP functions in postsynaptic muscles to constitutively provide a retrograde negative influence on synaptic strength. In light of these findings, a two-pronged scheme is proposed for the retrograde regulation of synaptic strength at the NMJ. On the one hand, a positive input from TOR is mediated through S6K/eIF4A and eIF4E to enhance postsynaptic translation. Synaptic compensation in GluRIIA mutant larva appears to rely mostly on this axis as evidenced by strong sensitivity to S6K heterozygosity and no change in the proportion of phosphorylated 4E-BP versus non-phosphorylated 4E-BP levels. Opposing this positive input, 4E-BP inhibits translation by sequestering eIF4E and adjusting the degree of retrograde compensation. Indeed, loss of 4E-BP leads to a strong increase in quantal content that is highly sensitive to eIF4E heterozygosity but not sensitive to S6K heterozygosity. The balance between these two forces reveals itself also when 4E-BP loss-of-function mutants are rescued by a non-phosphorylatable 4E-BP transgene. In this combination TOR can no longer inhibit 4E-BP, and this study finds that the presynaptic neurotransmitter release is lower than wild-type, similarly to what is observed in TOR hypomorphic mutants (Penney, 2012). A working model is proposed in which the negative force of 4E-BP is under constant check via phosphorylation by TOR, and the positive input from TOR/S6K is constitutively countered by 4E-BP’s ability to sequester eIF4E, a dynamic duel that ensures a tight regulation of synaptic strength (Kauwe, 2016).
The molecular mechanisms regulating animal tissue size during development are unclear. This question has been extensively studied in the Drosophila wing disc. Although cell growth is regulated by the kinase TORC1, no readout exists to visualize TORC1 activity in situ in Drosophila. Both the cell cycle and the morphogen Dpp are linked to tissue growth, but whether they regulate TORC1 activity is not known. This study developed an anti-phospho-dRpS6 antibody that detects TORC1 activity in situ. Unexpectedly, it was found that TORC1 activity in the wing disc is patchy. This is caused by elevated TORC1 activity at the cell cycle G1/S transition due to CycD/Cdk4 phosphorylating TSC1/2.TORC1 is also activated independently of CycD/Cdk4 when cells with different levels of Dpp signaling or Brinker protein are juxtaposed. This study has thereby characterize the spatial distribution of TORC1 activity in a developing organ (Romero-Pozuelo, 2017).
During animal development, tissues increase tremendously in mass, yet stop growing at very stereotyped sizes in a robust manner. For instance, the Drosophila wing is specified as a cluster of circa 50 cells, which increases in mass ~500-fold before terminating growth. Once growth has ceased, the left and right wings of an individual fly are virtually identical in size, to within 1%, illustrating the robustness of this process. How animal tissue size is regulated is a fundamental open question in developmental biology (Romero-Pozuelo, 2017).
As mitotically growing tissues develop, two independent cellular processes occur in a coordinated manner: proliferation and cell growth. By itself, proliferation -- the division of cells -- does not lead to mass accumulation. This was nicely shown in the Drosophila wing where overexpression of E2F speeds up the cell cycle, but leads to a normally sized tissue containing more, smaller cells. For a tissue to grow, cells need to accumulate biomass. The mechanisms interconnecting cell proliferation and cell growth are not completely understood. In organisms from yeast to humans, growth is in large part regulated by the target of rapamycin complex 1 (TORC1) kinase. TORC1 promotes biomass accumulation by promoting anabolic metabolic pathways such as protein, lipid, and nucleotide biosynthesis, while repressing catabolic processes such as autophagy. Hence, to understand tissue growth it would be of interest to study the spatial distribution of TORC1 activity in a developing tissue. This line of investigation has been hampered, however, by the lack of readouts for TORC1 activity that can be used in situ (Romero-Pozuelo, 2017).
One signaling pathway that strongly affects tissue size is the Dpp pathway. Dpp is expressed and secreted by a stripe of cells in the medial region of the wing imaginal disc, and forms an extracellular morphogen gradient that both helps to pattern the wing and affects its size. In the absence of Dpp signaling during development, only small rudimentary wings are formed. In contrast, overexpression of Dpp leads to strong tissue overgrowth, in particular along the axis of the morphogen gradient. Several models have been proposed for how Dpp signaling regulates wing size. The exact mechanism by which Dpp regulates tissue size, however, is an unresolved issue. Dpp signaling acts to repress expression of a transcription factor called Brinker. Brinker appears to mediate most of the size effects of Dpp signaling. When Brinker is genetically removed, Dpp signaling becomes dispensable for wing growth. Given that Dpp signaling promotes tissue growth, an open question is whether Dpp signaling promotes TORC1 activity (Romero-Pozuelo, 2017).
Thia study examined whether Dpp signaling promotes TORC1 activity in the Drosophila wing disc. To this end, a phospho-RpS6 (pS6) antibody was developed that allows TORC1 activity to be assayed in situ in tissue. This reagent reveals unexpectedly that TORC1 activity in the growing wing disc is neither uniform nor graded, but is instead patchy. This patchiness is mediated via CycD/Cdk4 and the tuberous sclerosis 1 (TSC1)-TSC2 complex in response to cell cycle stage. Using this pS6 antibody, this study found that TORC1 activity is also induced by discontinuities in Dpp signaling or discontinuities in Brinker levels. It is proposed that these discontinuous conditions may be analogous to regenerative conditions that happen in the wing disc in response to tissue damage. In sum, this work reveals the pattern of TORC1 activity in the context of a developing organ (Romero-Pozuelo, 2017).
TORC1 activity in the wing disc is modulated by the cell cycle, with cells in early S phase showing the highest TORC1 activity. Interestingly, an accompanying paper finds similar results in the Drosophila eye disc (Kim, 2017). This might reflect a metabolic requirement by early S-phase cells for large amounts of nucleotide biosynthesis, an anabolic process promoted by TORC1. Indeed, in various contexts S6K and TORC1 activity were found to be required for the transition from G1 to S. Connections between mechanistic TOR (mTOR) and the cell cycle have previously been found in cultured cells. In human fibroblasts, mTOR shuttles in and out of the nucleus in a cell cycle-dependent manner, peaking in the nucleus shortly before S phase. The relevance of this subcellular relocalization to what is observe in this study, however, is unclear. In fibroblasts, S6K1 activity was found to be highest during early G1, whereas in HeLa cells it was found to be highest during M phase. In sum, it is unclear to what extent cells in culture recapitulate endogenous development, or whether the influence of the cell cycle on TORC1 activity is very context dependent. The TSC1/2 complex has been reported to be phosphorylated by cell cycle-dependent kinases.TSC1 is phosphorylated on Thr417 by Cdk1 during the G2/M transition. This inhibitory phosphorylation would lead to elevated TORC1 activity during G2/M, which does not fit with what was observe here, and thus might be relevant in a different developmental context. Instead, this study found that TSC2 can be phosphorylated by the CycD/Cdk4 complex on Ser1046, and possibly other sites as well, and that this leads to activation of TORC1. This fits with several observations in the literature. Firstly, in U2OS cells the TSC complex was also found to bind cyclin D, leading to its phosphorylation at unknown sites. In U2OS cells, this causes destabilization of the Tsc1 and Tsc2 proteins, which was not observed in this study. Secondly, Tsc1/2 and CycD/Cdk4 were previously found to interact genetically in Drosophila: The reduced tissue growth caused by Tsc1 + Tsc2 overexpression was found to be fully suppressed by expression of CycD + Cdk4. This fits well with the current data suggesting that CycD/Cdk4 directly inhibits the TSC complex via phosphorylation. Thirdly, Cyclin D and Cdk4 were previously reported in Drosophila to promote cell and tissue growth, fitting with activation of the TORC1 complex by CycD/Cdk4. It is worth noting that some patchy TORC1 activity is still seen in CycD- or Cdk4-null discs and in discs with the single phospho-site mutations in TSC2. Hence it is possible that Cdk4 may not be the only factor regulating TORC1 activity in response to the cell cycle, and that Cdk4 might phosphorylate TSC2 on additional sites (Romero-Pozuelo, 2017).
What are the roles of CycD/Cdk4 in cell cycle progression and cell growth? Whereas mammals have three cyclin D genes, CycD1-3, and two CycD binding kinases, Cdk4 and Cdk6, Drosophila has a single CycD, a single Cdk4, and no Cdk6. Hence Drosophila provides an opportunity to elucidate the function of the CycD/Cdk4 complex without difficulties arising from redundancy. Indeed, results in Drosophila clearly show that CycD/Cdk4 promotes cell growth and not cell cycle progression. Both CycD- and Cdk4-null animals are viable, and fluorescence-activated cell sorting (FACS) analysis of null cells revealed that they have a normal cell cycle profile, indicating that they are dispensable for normal cell cycle progression. Instead, Cdk4- and CycD-null animals are 10%-20% smaller than controls, indicating that they promote cell growth. The finding that CycD/Cdk4 activates TORC1 during the G1/S transition can provide one mechanism by which the CycD/Cdk4 complex promotes growth. Hence, from these data it is proposed that in Drosophila the CycD/Cdk4 complex is not part of the core machinery required for cell cycling, but is rather an effector 'side branch' activated at G1/S to promote cell growth. Data from the mouse suggest something similar. CycD1, CycD2, and CycD3 knockout mice are all viable. One could imagine this to be due to redundancy between these three genes, but actually CycD1, CycD2, CycD3 triple-knockout mice survive to mid-gestation, and the triple-knockout mouse embryonic fibroblasts proliferate relatively normally. The mid-gestation lethality of the triple knockouts appears to be due to specific effects in hematopoietic and myocardial cells. Hence, cyclins D1-D3 are also dispensable for cell cycle progression in mice. Interestingly, CycD1 knockout mice and CycD1, CycD2 double-knockout mice are viable but have reduced body size, reminiscent of the size phenotype observed in CycD knockout flies. In sum, despite CycD/Cdk4 being claimed in most reviews on the cell cycle as playing an important role in G1/S progression, it appears that this complex may function rather to promote cell growth in a cell cycle-dependent manner (Romero-Pozuelo, 2017).
Does Dpp control growth in the wing? When discontinuities in Dpp activity or in Brinker levels were genetically induce, activation was observed of TORC1 at the site of discontinuity. Hence, Dpp signaling per se does not appear to activate TORC1; rather, the comparison between high Dpp signaling and low Dpp signaling cells does. In an unperturbed disc, no pattern of pS6 staining was observed that correlates with the Dpp activity gradient, which is highest medially and drops toward the anterior and posterior extremities. This might be due to the fact that in an unperturbed disc the Dpp and Brinker gradients are smooth and do not have such discontinuities. A similar effect of Dpp was previously observed on cell prolife ration, except that in this case the effect of the Dpp discontinuity was very transient, lasting only a few hours after clone induction, whereas the effect seen on growth is sustained. Dpp signaling is, nonetheless, required for growth, because in the absence of Dpp, small vestigial wings are formed. Hence one interpretation might be that low levels of Dpp signaling are continuously required for growth, but that Dpp signaling becomes instructive for tissue growth only when discontinuities in the gradient arise, perhaps as a result of tissue damage or cell delamination, to initiate a regenerative response (Romero-Pozuelo, 2017).
One additional interesting non-autonomous phenomenon observed is that sometimes when a region of the wing disc has high pS6 levels, the rest of the disc loses its typically patchy pS6 pattern and becomes pS6 negative. This phenomenon is not understood, and future work will be necessary to understand it molecularly (Romero-Pozuelo, 2017).
The mechanistic (or mammalian) target of rapamycin complex 1 (mTORC1) controls cell growth, proliferation, and metabolism in response to diverse stimuli. Two major parallel pathways are implicated in mTORC1 regulation including a growth factor-responsive pathway mediated via TSC2/Rheb and an amino acid-responsive pathway mediated via the Rag GTPases. This study identified and characterize three highly conserved growth factor-responsive phosphorylation sites on RagC, a component of the Rag heterodimer, implicating cross talk between amino acid and growth factor-mediated regulation of mTORC1. RagC phosphorylation is associated with destabilization of mTORC1 and is essential for both growth factor and amino acid-induced mTORC1 activation. Functionally, RagC phosphorylation suppresses starvation-induced autophagy, and genetic studies in Drosophila reveal that RagC phosphorylation plays an essential role in regulation of cell growth. Finally, mTORC1 was identified as the upstream kinase of RagC on S21. These data highlight the importance of RagC phosphorylation in its function and identify a previously unappreciated auto-regulatory mechanism of mTORC1 activity (Yang, 2018).
mTOR is an evolutionarily conserved atypical serine/threonine kinase belonging to the phosphoinositide 3 kinase (PI3K)-related kinase family. mTOR is found in two structurally and functionally distinct complexes-mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2)-defined by their unique components, in particular raptor (mTORC1) and rictor (mTORC2). Through the coordinated phosphorylation of its downstream effectors, mTORC1 integrates extra- and intra-cellular signal inputs such as amino acids, growth factors (GF), stress, and energy status, to regulate major cellular processes including growth, proliferation, and survival. Underlining its crucial role in cellular and organismal homeostasis, mTORC1 dysregulation occurs in numerous human diseases including cancer, metabolic disorders, and neurodegeneration. Growth factors and amino acids both acutely enhance mTORC1 activity, and two different types of small GTPases-Ras-homolog enriched in brain (Rheb) and the Rag GTPases-cooperatively regulate mTORC1 activity via these two parallel activation mechanisms. Rheb is activated under conditions of high cellular ATP and upstream growth factor signals. Once activated, Rheb interacts with and activates mTORC1 and is required for mTORC1 activation by all signals, including amino acids. Rag GTPases are considered amino acid-specific regulators of the mTORC1 pathway. Mammals have four Rag proteins-RagA to RagD-which form obligate heterodimers comprising RagA or RagB together with RagC or RagD. Amino acids cause Rag GTPases to switch to an active conformation, in which RagA/B is GTP-loaded and Rag C/D is GDP-loaded. The active Rag heterodimer physically interacts with raptor, recruiting mTORC1 to the lysosome where its activator Rheb resides. Extensive work has revealed several mechanisms implicated in the regulation of Rag activity that enables them to function as nutrient sensors. A common feature among these is the control of Rag nucleotide status, particularly through the activation of guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). These include the Ragulator (GEF for Rag A/B; Bar-Peled, 2012), the GAP Activity Towards Rags complex 1 (GATOR1) complex (GAP for RagA/B; Bar-Peled, 2013), folliculin (FLCN, GAP for RagC/D; Petit, 2013; Tsun, 2013), and leucyl-tRNA synthetase (LeuRS, GAP for RagD; Han, 2012). Ubiquitination has also recently emerged as a post-translational modification (PTM) capable of inhibiting Rag GTPase signaling by recruiting GATOR1 to RagA. Importantly, these pathways regulating Rag activity are all amino acid-dependent, and much less is known about the control of growth factor-mediated Rag GTPase signaling (Yang, 2018).
In a recent global mass spectrometry-based phosphoproteomics study in adipocytes, insulin-dependent phosphorylation was observed of several highly conserved residues on RagC including S2, S21, and T394. These data highlight a possible role for the Rag GTPases in mTORC1 growth factor sensing. This study demonstrates that both growth factors and amino acids trigger RagC phosphorylation and that phosphorylated RagC potentiates mTORC1 activity and affects mTORC1-dependent cell growth and autophagy. Moreover, the phosphorylation of RagC at S21 (and likely T394) was shown to be catalyzed directly by mTORC1, revealing a novel auto-regulatory feedback loop within the mTORC1 signaling pathway (Yang, 2018).
This study identified a new auto-regulatory branch of mTORC1 signaling, involving phosphorylation of the Rag GTPase RagC. This is the first report that Rag GTPase phosphorylation can regulate mTORC1 activity. More importantly, the results confirm that Rag GTPases are not only involved in the amino acid-sensing mTORC1 pathway, but could also participate in growth factor sensing in the mTORC1 pathway. Although previous studies show that in Rag heterodimers, the GTP/GDP loading of Rag heterodimers plays a dominant role in the interaction between Rag heterodimers and mTORC1, the data indicate that RagC is also a positive regulator of mTORC1 through post-translational modification. Interestingly, phosphoproteomics data suggest that most phosphorylation is concentrated on RagC compared with other Rag GTPases, and S21 is not conserved between RagC and RagD, suggesting that RagC is not functionally redundant and potentially has distinct biological functions to RagD (Yang, 2018).
One of the RagC phosphorylation sites, S21, was established as a novel rapamycin-insensitive mTORC1 substrate in vitro and in cells, and the T394 is phosphorylated by mTOR in vitro. The S2 and T394 sites may also be mTORC1 substrates in vivo, because the kinetics of their phosphorylation resembles that of other bona fide mTORC1 substrates and they also have surrounding sequence features matching the preferred sequence motif of mTORC1. These findings indicate the presence of a positive feedback loop between mTORC1 and RagC, which may contribute to the fine-tuning of mTORC1 activity (Yang, 2018).
There is evidence that the stability of the raptor-mTOR complex is related to mTORC activity, and the current data implicate RagC phosphorylation in the destabilization of mTORC1. This is likely to be a direct effect of RagC phosphorylation, because RagC 3E still destabilized mTOR-raptor complex under serum starvation. This is consistent with the observation that RagC 3E causes hyper-phosphorylation of ULK1 and inhibits autophagy under serum starvation. The next major question is what is the underlying cause of this instability. One possibility is that RagC phosphorylation influences the interaction with other regulators, resulting in 'locking' or 'opening' of the mTOR-raptor complex. Interestingly, it was observed that RagC 3A binds more FLCN, which is a GAP for RagC/D, and RagC 3E can bind more raptor under both steady and amino acid starvation/re-fed condition. One possibility is that RagC phosphorylation regulates its nucleotide binding status by modulating the interaction with FLCN. However, no substantial difference was observed in FLCN binding between wild-type RagC and RagC 3E, or raptor binding between wild-type RagC and 3A. The temporal change in mTOR/raptor stability upon amino acid re-feeding is similar to that of FLCN/RagC stability, but not with raptor/RagC interaction. A possible explanation is that FLCN has two functions: serving as a GAP for RagC/D and a 'lock' for mTORC1. This model could help explain why FLCN releases from Rag GTPases in the presence of amino acids if it is a GAP for RagC/D, which is a positive regulator for mTORC1 activity: After activating RagC/D, FLCN needs to be disassociated from the lysosome to unlock mTORC1, and RagC phosphorylation may affect this process. Further studies will be needed to investigate these possibilities (Yang, 2018).
Other explanations cannot be ruled out for the impact of RagC phosphorylation on impaired mTORC1 activity. For example, it is well established that raptor recruits substrate proteins such as S6K and 4E-BP1 to mTORC1 so that they can be phosphorylated by mTOR. Therefore, RagC phosphorylation may affect the recruitment of mTORC1 substrates by raptor. Recently, two elegant studies showed that under amino acid or growth factor starvation, the Rag heterodimer binds and recruits TSC2 to lysosomes to inhibit Rheb, resulting in mTORC1 inactivation. Therefore, a final possibility is that RagC phosphorylation may mediate its effects by acting through TSC2. Future studies into the underlying mechanics of how RagC phosphorylation exerts its effects on mTORC1 signaling are therefore likely to shed light on this newly identified mechanism that sits at the intersection between amino acid sensing and growth factor signaling (Yang, 2018).
mTOR Complex (mTORC1) promotes cell growth and proliferation in response to nutrients and growth factors. Amino acids induce lysosomal translocation of mTORC via the Rag GTPases. Growth factors activate Ras homolog enriched in brain (Rheb), which in turn, activates mTORC at the lysosome. Amino acids and growth factors also induce the phospholipase D (PLD)-phosphatidic acid (PA) pathway, required for mTORC signaling through mechanisms that are not fully understood. Using human and murine cell lines, along with immunofluorescence, confocal microscopy, endocytosis, PLD activity, and cell viability assays, this study shows that exogenously supplied PA vesicles deliver mTORC to the lysosome in the absence of amino acids, Rag GTPases, growth factors, and Rheb. Of note, pharmacological or genetic inhibition of endogenous PLD prevented mTORC lysosomal translocation. This study observed that precancerous cells with constitutive Rheb activation through loss of TSC complex subunit (TSC2) exploit the PLD-PA pathway and thereby sustain mTORC activation at the lysosome in the absence of amino acids. These findings indicate that sequential inputs from amino acids and growth factors trigger PA production required for mTORC translocation and activation at the lysosome (Frias, 2019).
mTORC18 is a conserved serine/threonine catalytic complex that integrates signals from nutrients and growth factors to regulate cell growth, proliferation, survival, and metabolism. Activation of mTORC1 is a two-step process whereby amino acids induce Rag-dependent translocation of mTORC1 from the cytoplasm to the lysosome, followed by mTOR kinase activation by the lysosomal small GTPase Rheb upon growth factor stimulation (Frias, 2019).
Phospholipase D (PLD) and its product, the signaling lipid phosphatidic acid (PA) play a role in mTORC1 activation in response to amino acids and growth factors. Amino acids induce lysosomal translocation of PLD1. Once on the lysosome, PLD1 binds to Rheb, which activates PLD1 in response to growth factors. PLD1 is widely expressed in mammals and converts the most abundant membrane phospholipid phosphatidylcholine to choline and PA. Conserved basic amino acids in the FKBP12-rapamycin binding (FRB) domain of mTOR lead to proton dissociation to generate PA with two negative charges. This locks mTOR onto deprotonated PA, promoting mTORC1 assembly and stability (Frias, 2019).
This study reports that PA with an unsaturated fatty acid stimulates lysosomal translocation and activation of mTORC1 in the absence of amino acids, Rag GTPases, growth factors, or Rheb. This work provides a unifying model showing that PA is critical for translocation and full activation of mTORC1 at the lysosome in response to sequential signals provided by amino acids and growth factors (Frias, 2019).
The data support a model where PLD1, similar to mTORC1, acts like a coincidence detector and effector of both amino acids and growth factors. Amino acids induce PLD activity and production of PA. Exogenously supplied PA vesicles enter the cell through endocytosis and drive mTOR to the lysosome, suggesting that amino acids induce production of PA-containing endosomes that carry mTOR to the lysosome. In agreement, inhibition of endogenous PLD prevented mTOR translocation to the lysosome in response to amino acids. Amino acid-induced RagA/B-GTP RagC/D-GDP heterodimers provide a parallel pathway that locks mTOR on the lysosome. Amino acids also induce the translocation of PLD1 from cytoplasmic puncta to the lysosome. Once on the lysosome, PLD1 binds to Rheb. Growth factors activate Rheb, which then activates PLD1. Lysosomal PA production promotes further binding of PA to mTOR to allow complex stability and activation (Frias, 2019).
Previous studies showed that exogenously supplied PA induced mTORC1 activation in the presence but not in the absence of amino acids. This study was able to induce mTORC1 translocation to the lysosome and mTORC1 activity with exogenously supplied PA-18:1 vesicles in the absence of amino acids. The main difference between the two studies is that this study performed amino acid and serum deprivation (to prevent contamination of amino acids present in serum) for 1 h, followed by amino acid stimulation for 10 min. In contrast, the previous study performed amino acid starvation for 2 h after overnight serum starvation, followed by amino acid stimulation for 30 min (Frias, 2019).
Previous findings suggest that mTORC1 assembles before reaching the lysosome because binding of mTOR to the Rag GTPases requires the mTORC1 component raptor. PA promotes mTORC1 assembly and stability. PA-containing endosomes carrying mTORC1 to the lysosome is therefore an attractive model in which PA would allow mTORC1 formation and stability. This study showed that exogenously supplied PA can drive mTOR to the lysosome and induce mTORC1 activity in TSC2-null MEFs where RagC and D were genetically ablated. Therefore, it is proposed that delivery of mTORC1 to the lysosome does not require the Rags. However, this study found that residual retention of mTOR at the lysosome in TSC2-null MEFs was lost upon RagC and D knockdown. This suggests that the Rags operate in parallel to PA to lock mTORC1 on the lysosome, after mTORC1 delivery by PA. This study showed that genetic ablation of PLD1 induced lysosomal scattering. This favors the idea that PA-containing endosomes carry mTORC1 to the lysosome, fuse with the lysosome, and increase its size (Frias, 2019).
Exogenously supplied PA vesicles induce mTORC1 translocation and activation in the absence of Rheb, indicating that the key step in Rheb activation of mTORC1 is increased PLD1 activity and PA production. Consistent with this observation, the Rheb association with mTOR is independent of GTP loading, whereas the Rheb association with PLD1 depends on GTP loading. Additionally, this study found that PLD1/2 inhibitors, in combination, abolished mTORC1 activity in RagAGTP/GTP MEFs, indicating that PA production is required for complex assembly and stability. If mTORC1 is not intact, then constitutive Rag activation is lost. Thus, the effect of PA on mTORC1 is downstream of Rheb and parallel to Rag GTPases.
Genetic deletion of mTOR in mice is embryonic lethal. Unlike mTOR, mice with genetic deletion of PLD1, PLD2, or both are viable, suggesting that PLD and PA may not be required for steady state but rather acute activation of mTOR. PLD inhibitors preferentially killed TSC-null MEFs, whereas rapamycin did not. This suggests an advantage in terms of cancer therapeutics because targeting PLD may selectively target cancer cells, with minimal side effects. PLD inhibitors were developed from halopemide, a psychotropic drug extensively used in humans without toxicities. Thus, PLD inhibition might be a viable alternative to current therapies in cancers with mTORC1 hyperactivation that requires PLD-generated PA (Frias, 2019).
The TORC regulator GATOR1/SEACIT controls meiotic entry and early meiotic events in yeast. However, how metabolic pathways influence meiotic progression in metazoans remains poorly understood. This study examined the role of the TORC regulators GATOR1 and GATOR in the response to meiotic double-stranded breaks (DSB) during Drosophila oogenesis. In mutants of the GATOR component mio, meiotic DSBs trigger the constitutive downregulation of TORC activity and a permanent arrest in oocyte growth. Conversely, in GATOR mutants, high TORC activity results in the delayed repair of meiotic DSBs and the hyperactivation of p53. Unexpectedly, it was found that GATOR inhibits retrotransposon expression in the presence of meiotic DSBs in a pathway that functions in parallel to p53. Thus, these studies have revealed a link between oocyte metabolism, the repair of meiotic DSBs and retrotransposon expression (Wei, 2019).
Metabolism impacts meiotic progression during oogenesis. Target of Rapamycin Complex 1 (TORC1) is a multi-protein complex that functions as a master regulator of metabolism. In the presence of adequate nutrients and positive upstream growth signals, TORC1, which contains the serine/threonine kinase Target of Rapamycin, becomes active and functions to stimulate growth and inhibit catabolic metabolism through the phosphorylation of down-stream effector proteins. The Seh1 Associated Complex Inhibits TORC1 (SEACIT), originally identified in yeast, inhibits TORC1 activity in response to amino acid limitation. SEACIT, known as the GAP Activity Towards Rags complex 1 (GATOR1) in metazoans, is comprised of three highly conserved proteins Npr2/Nprl2, Npr3/Nprl3 and Iml1/Depdc5. In Drosophila and mammals, depleting any of the three GATOR1 components results in increased TORC1 activity and growth, as well as a reduced response to amino acid starvation. Thus, the role of the SEACIT/GATOR1 complex in the regulation of TORC1 activity is highly conserved in eukaryotes (Wei, 2019).
The multi-protein GATOR2 complex, known as Seh1 Associated Complex Activates TORC1 (SEACAT) in yeast, inhibits the activity of GATOR1 and thus functions to activate TORC1 (see Mio prevents the constitutive downregulation of TORC1 activity in response to meiotic DSBs). In metazoans, the GATOR2 complex functions in multiple amino acid sensing pathways. In tissue culture cells, depleting GATOR2 components results in the constitutive activation of GATOR1 and the permanent downregulation of TORC1 activity. However, genetic studies of the role of individual GATOR2 components in Drosophila, indicate that the requirement for the GATOR2 complex is more nuanced when examined in the context of a multicellular animal. For example, mutations in the GATOR2 component mio, result in a block to oocyte growth and differentiation, due to the constitutive downregulation of TORC1 activity in the female germline. However, mio is not required to maintain TORC1 activity in most somatic tissues of Drosophila (Wei et al., 2016). Why there is a tissue specific requirement for mio in the female germline of Drosophila is currently unknown (Wei, 2019).
In single celled eukaryotes, nutrient limitation often facilitates meiotic entry. In the yeast Saccharomyces cerevisiae, the down-regulation of TORC1 by SEACIT/GATOR1 in response to amino acid stress promotes both meiotic entry and early meiotic progression. Surprisingly, as is observed in yeast, during Drosophila oogenesis the GATOR1 complex promotes meiotic entry. These data raise the intriguing possibility that in Drosophila the GATOR1 complex and low TORC1 activity may be critical to the regulation of additional events of the early meiotic cycle (Wei, 2019).
This study reports that the GATOR complex is critical to the response to meiotic DSB during Drosophila oogenesis. Restraining TORC1 activity via a pathway that involves both GATOR1 and the Tuberous sclerosis complex (TSC) promotes the timely repair of meiotic DSBs and prevents the hyperactivation of p53 in the female germline. Notably, the delayed repair of meiotic DSBs in GATOR1 mutants is due, at least in part, to the hyperactivation of the TORC1 target S6K. Conversely, the data indicate that the GATOR2 component Mio opposes the activity of GATOR1 in the female germline, thus preventing the constitutive downregulation of TORC1 activity and allowing for the growth and development of the oocyte in later stages of oogenesis. Thus, this study has identified a regulatory loop required to modulate TORC1 activity in response to meiotic DSBs during Drosophila oogenesis. Finally, during the course of these studies, it was observed that the GATOR1 complex prevents the derepression of retrotransposon expression in the presence of meiotic DSBs (Wei, 2019).
Previous work has shown that in Drosophila, mutations in the GATOR2 component mio, result in the constitutive activation of the GATOR1 pathway in the female germline but not in somatic tissues. This study demonstrates that the tissue specific requirement for mio during oogenesis is due, at least in part, to the generation of meiotic DBSs during oogenesis. In Drosophila, only the female germline undergoes meiotic recombination and thus experiences the genotoxic stress associated with developmentally programmed DSBs. This study shows that in mio mutants, blocking the formation of meiotic DSBs prevents the constitutive downregulation of TORC1 activity thus allowing for the growth and development of the oocyte. These data are consistent with the model that meiotic DSBs trigger the activation of a TORC1 inhibitory pathway that must be opposed and/or attenuated by the GATOR2 component Mio (see A working model for the role of the GATOR complex in the response to meiotic DSBs) (Wei, 2019).
While there are several possible models that might explain these data, it is believed the most parsimonious explanation for these results is that the TORC1 inhibitory pathway activated by meiotic DSBs, involves both GATOR1 and TSC. This model is consistent with the ability of both GATOR1 and TSC depletions to rescue the mio mutant phenotype. Additionally, recent reports indicate that GATOR1 and TSC act in a common pathway to downregulate TORC1 activity in response to multiple upstream inhibitory inputs. Previously, it was determined that in Drosophila, amino acid starvation induces a dramatic GATOR1/TSC dependent decrease in TORC1 activity in somatic tissues, that far exceeds any reduction in TORC1 activity observed in GATOR2 null mutants. This observation strongly suggests that, in addition to the removal of the GATOR2 inhibition of GATOR1, there is an activation step that is required to fully potentiate the GATOR1/TSC pathway (Wei, 2019).
Thus, based on these data the following model is proposed. Meiotic DSBs activate, or are required to maintain, a GATOR1/TSC dependent pathway that downregulates TORC1 activity in the female germline. The GATOR2 component Mio is required to oppose or turnoff this pathway to prevent the constitutive downregulation of TORC1 activity in later stages of oogenesis. While it is believed that the data support the role of the GATOR1/TSC pathway, it is conceded that an alternative regulator of TORC1 activity may also be critical to the downregulation of TORC1 activity in response to meiotic DSBs (Wei, 2019).
Hyperactivation of TORC1 has been linked to defects in the DNA damage response in single celled and multicellular organisms. The observation that meiotic DSBs likely promote the GATOR1 dependent downregulation of TORC1 activity during Drosophila oogenesis, suggested that limiting TORC1 activity may be important to the regulation of meiotic DSB repair. In previous work, it was found that GATOR1 mutant ovaries had TORC1 activity levels approximately three times higher than those observed in wild-type ovaries. This study demonstrates that GATOR1 mutant ovaries exhibit multiple phenotypes consistent with the misregulation of meiotic DSB repair including, an increase in the steady state number of Mei-W68/Spo-11 induced DSBs, the retention of meiotic DSBs into later stages of oogenesis and the hyperactivation of p53. Importantly, RNAi depletions of Tsc1 partially phenocopied the GATOR1 ovarian defects. Thus, the misregulation of meiotic DSBs observed in GATOR1 mutant oocytes are due to high TORC1 activity and not to a TORC1 independent function of the GATOR1 complex (Wei, 2019).
Epistasis analysis between the GATOR1 component nprl3 and the Rad51 homolog spnA, strongly suggest that GATOR1 impacts the repair, rather than the generation, of meiotic DSBs. This study determined that double mutants of nprl2 and the Rad51 homolog spnA, which is required for the repair of meiotic DSBs, have approximately the same number of DSBs as spnA single mutants. These data are consistent with GATOR1 and spnA influencing the common process of DNA repair and are inconsistent with GATOR1 mutants producing supernumerary breaks (Wei, 2019).
These observations on the role of the GATOR1 complex during Drosophila oogenesis are particularly intriguing in light of similar meiotic defects observed in a npr3 mutants in Saccharomyces cerevisiae. In the sporulation proficient strain SK1, npr3 mutant cells enter meiosis and express the transcription factor and master regulator of gametogenesis IME1 with wild-type kinetics. Subsequently, npr3 mutants exhibit a mild delay in the generation of meiotic DNA breaks, but a substantial delay in the repair of meiotic DSBs. Thus, yeast and Drosophila SEACIT/GATOR1 mutants share a common meiotic phenotype, the delayed repair of meiotic DSBs. These results raise the intriguing possibility that low TORC1 activity may be a common feature of the early meiotic cycle in many organisms (Wei, 2019).
Notably, the data indicate that the delay in the repair of meiotic DSBs in GATOR1 mutants is due to the hyperactivation of the TORC1 downstream target S6K. S6K is a critical downstream effector of TORC1 that impacts multiple essential cellular processes including, but not limited to cell growth, energy balance and aging. Intriguingly, in mammals, S6K has been implicated in the regulation of the DNA damage response with hyperactivation of the TORC1-S6K pathway resulting in the accumulation of unrepaired DSBs and genome instability. Thus, similar to what is reported in mammals, the data are consistent with the model that the hyperactivity of the TORC1/S6K axis delays the repair of DSBs in Drosophila (Wei, 2019).
Finally, it was determined that GATOR1 mutants have a diminished response to DSBs outside the female germline in somatic tissues of Drosophila. Similar to what is observed in TSC mutant cells in humans that have increased levels of TORC1 activity, this study find that GATOR1 mutant embryos have a reduced ability to survive low levels of γ-irradiation. Moreover, in the somatic follicle cells of the ovary a delay was observed in the repair of DSBs after adult females are exposed to low levels of γ-irradiation. Thus, in Drosophila inappropriately high TORC1 activity delays the repair of DSBs in both the germline and somatic tissues (Wei, 2019).
The initiation of homologous recombination through the programmed generation of DNA double-stranded breaks (DSBs) is a universal feature of meiosis. DSBs represent a dangerous form of DNA damage that can result in dramatic and permanent changes to the germline genome. To minimize this destructive potential, the generation and repair of meiotic DSBs is tightly controlled in space and time . The activation of transposable elements represents an additional threat to genome integrity in germ line cells. Genotoxic stress, resulting from DNA damage, has been implicated in the deregulation of transposons in multiple organisms. Thus, germ line cells may be at an increased risk for transposon derepression due to the genotoxic stress associated with meiotic recombination. Consistent with this hypothesis, germ line cells have evolved extensive surveillance systems to detect and silence transposons beyond the pathways present in most somatic tissues (Wei, 2019).
Previous studies have shown that DNA damage promotes the deregulation of retrotransposon in multiple organisms, including Drosophila. In line with these studies, this study found that in GATOR1 mutants, the DSBs that initiate meiotic recombination trigger the deregulation of retrotransposon expression. Similarly, p53 mutant females derepress retrotransposon expression during oogenesis, but as observed in GATOR1 mutants, primarily in the presence of meiotic DSBs. Double mutants of nprl3, p53 exhibit a dramatic increase in retrotransposon expression relative to either p53 or nprl3 single mutants, implying that p53 and GATOR1 act through independent pathways to repress retrotransposon expression in the female germline. One possibility is that both GATOR1 and p53 independently impact genome stability. Thus, disabling both pathways may have an additive effect on both genome stability and retrotransposon expression. Consistent with the hypothesis that genome instability drives retrotransposon expression, this study found that mutants in spnA/Rad51, which fail to repair meiotic DSBs, also exhibit increased transcription of multiple retrotransposons. Intriguingly, the SpnA homolog Rad51, as well as other genes required for DNA repair, was recently identified in a high throughput screen for genes that suppress (Long Interspersed Element-1) LINE1 expression in mammalian tissue culture cells (Wei, 2019).
However, the current data also suggest that the GATOR1 complex may influence retrotransposon expression independent of the regulation of TORC1 activity. While both GATOR1 and TSC are required for the efficient repair of meiotic DSBs, in contrast to GATOR1 mutant ovaries, little to no increase was observed in retrotransposon expression in the Tsc1 depleted ovaries. We believe this reflects the incomplete depletion of Tsc1 by RNAi resulting in a reduced retention of meiotic DSBs relative to GATOR1 mutants. However, a second possibility is that the GATOR1 complex inhibits retrotransposon expression independent of TORC1 inhibition. As is observed with spnA the depletion of GATOR1 components, but not TSC components result in the activation of LINE1 expression in HeLa cells. Taken together, these data hint that the GATOR1 complex may impact retrotransposon expression in the germline via two independent pathways: First by promoting the repair of meiotic DSBs through the downregulation of TORC1 activity and second via a pathway that functions independent of TORC1 inhibition (Wei, 2019).
Genes encoding components of the GATOR1 complex are often deleted in cancers. As is observed in GATOR1 mutants, cancer cells frequently have increased TORC1 activity, increased genomic instability and increased retrotransposon expression. Thus, in the future it will be important to identify the molecular mechanism by which the GATOR1 complex influences both the response to genotoxic stress and the expression of retrotransposons under both normal and pathological conditions (Wei, 2019).
This study shows that the dietary lipid cholesterol, which is required as a component of cell membranes and as a substrate for steroid biosynthesis, also governs body growth and maturation in Drosophila via promoting the expression and release of insulin-like peptides. This nutritional input acts via the nutrient sensor TOR, which is regulated by the Niemann-Pick-type-C 1 (Npc1) cholesterol transporter, in the glia of the blood-brain barrier and cells of the adipose tissue to remotely drive systemic insulin signaling and body growth. Furthermore, increasing intracellular cholesterol levels in the steroid-producing prothoracic gland strongly promotes endoreduplication, leading to an accelerated attainment of a nutritional checkpoint that normally ensures that animals do not initiate maturation prematurely. These findings, therefore, show that a Npc1-TOR signaling system couples the sensing of the lipid cholesterol with cellular and systemic growth control and maturational timing, which may help explain both the link between cholesterol and cancer as well as the connection between body fat (obesity) and early puberty (Texada, 2022).
Nutrition is one of the most important influences on developmental growth and maturation. Malnutrition or disease can impair growth and delay puberty, whereas obese children enter puberty early. Similarly, Drosophila larvae exposed to poor nutrition, tissue damage, or inflammation delay their development, whereas rich conditions promote rapid growth and maturation. These environmental factors are coupled to the appropriate gating of steroid production via internal checkpoints, one of which is a nutrition-dependent “critical weight” (CW) required to initiate the maturation process (Texada, 2022).
This suggests that signals reflecting nutritional state and body-fat storage play a key role in activating the neuroendocrine pathways that trigger puberty. Although studies suggest that the adipokine leptin may be involved, the mechanisms linking body fat to puberty are poorly defined, and the potential involvement of lifestyles associated with excessive accumulation of cholesterol, one of the most important lipids, has not been considered. In humans, white adipose tissue is the main site of cholesterol storage and can contain over half the body's total cholesterol in obesity (Texada, 2022).
The results show that dietary cholesterol intake promotes systemic body growth through insulin-dependent pathways and that animals raised on high dietary cholesterol initiate maturation earlier. It was show nthat cholesterol is sensed through an Npc1-regulated TOR-mediated mechanism in the fat body and the glial cells of the BBB, which relay information to the IPCs within the brain to promote insulin expression and release, thus coupling growth and maturation with cholesterol status (Texada, 2022).
Insect CW likely evolved as a mechanism ensuring that maturation will not occur unless the animal has accumulated adequate nutrient stores to survive the nonfeeding metamorphosis period and has completed sufficient growth to produce an adult of proper size and thus of maximal fitness. Likewise, the link between body fat and maturation in humans probably ensures adequate stores of fat before maturation onset to support pregnancy and reproductive success (Texada, 2022).
In Drosophila, insulin signaling plays a critical role in coordinating steroidogenesis with nutritional conditions. Insulin acts upon the prothoracic gland (PG) and induces a small ecdysone peak early in L3 that is correlated with CW attainment. In combination with nutrition-related signaling mediated via insulin, nutrient availability is also assessed directly in the PG and is coupled to irreversible endoreduplication that permits ecdysone production at CW (Texada, 2022).
These findings show that accumulation of cholesterol in the PG, induced by the loss of Npc1a, drives a remarkable TOR-dependent increase in endoreduplication and leads to inappropriate attainment of CW. Taken together, these findings indicate that cholesterol sensed by the BBB glia and the fat body promotes growth through insulin signaling and that cholesterol sensed by the PG accelerates maturation through ecdysone signaling.
Loss of Npc1 function in humans leads to the Niemann-Pick lysosomal storage disorder, marked by intracellular cholesterol accumulation. Although neurodegeneration is the hallmark of NPC disease, including in a Drosophila model,70
alterations in glial, adipose, hepatic, and endocrine systems are also components of NPC syndrome. In humans, Npc1 itself is strongly expressed in glia and in adipose tissues, especially in obese individuals, and variants in Npc1 are associated with obesity, type-2 diabetes, and hepatic lipid dysfunction (Texada, 2022).
These findings link glial and adipose-tissue cholesterol sensing through Npc1 to systemic growth and metabolic control through effects on insulin signaling. It was also found that intracellular cholesterol accumulation driven by Npc1 loss leads to hyperactivation of TOR that drives increases in DNA replication and cell growth. TOR activity is frequently upregulated in cancer, and the results therefore provide mechanistic insight for understanding the emerging link between cholesterol and a range of cancers.
As the coupling of nutrition with growth and maturation is ancient and highly conserved, this work provides a foundation for understanding how cholesterol is coupled to developmental growth and maturation initiation in humans. These findings link a high concentration of this particular lipid in adipose tissues to the neuroendocrine initiation of maturation, which may explain the critical link between obesity (body fat) and early puberty (Texada, 2022).
Organisms modulate their growth according to nutrient availability. Although individual cells in a multicellular animal may respond directly to nutrient levels, growth of the entire organism needs to be coordinated. This study provides evidence that in Drosophila, coordination of organismal growth originates from the fat body, an insect organ that retains endocrine and storage functions of the vertebrate liver. A genetic screen for growth modifiers discovered slimfast, a gene that encodes an amino acid transporter. Remarkably, downregulation of slimfast specifically within the fat body causes a global growth defect similar to that seen in Drosophila raised under poor nutritional conditions. This involves TSC/TOR signaling in the fat body, and a remote inhibition of organismal growth via local repression of PI3-kinase signaling in peripheral tissues. These results demonstrate that the fat body functions as a nutrient sensor that restricts global growth through a humoral mechanism (Colombani, 2003).
In multicellular organisms, the control of growth depends on the integration of various genetic and environmental cues. Nutrient availability is one of the major environmental signals influencing growth and, as such, has dictated adaptative responses during evolution toward multicellularity. In particular, complex humoral responses ensure that growth and development are properly coordinated with nutritional conditions (Colombani, 2003).
In isolated cells, amino acid withdrawal leads to an immediate suppression of protein synthesis, suggesting that cells are protected by active sensing mechanims that block translation prior to depletion of internal amino acid stores. In many mammalian cell types, changes in amino acid diet affect the binding of the translation repressor 4EBP1 to initiation factor eIF4E and the activity of ribosomal protein S6 kinase (S6K). These two signaling events require the activity of TOR (target of rapamycin), a conserved kinase recently shown to participate in a nutrient-sensitive complex both in mammalian cells and in yeast. Mutations in the Drosophila TOR homolog (dTOR) results in cellular and physiological responses characteristic of amino acid deprivation and establish that TOR is cell autonomously required for growth in a multicellular organism. Furthermore, the TSC (tuberous sclerosis complex) tumor suppressor, consisting of a TSC1 and TSC2 heterodimer (TSC1/2), as well as the small GTPase Rheb participate to the regulation of TOR function. Overall, these data suggest that TSC, Rheb, TOR, and S6K participate in a conserved pathway that coordinates growth with nutrition in a cell-intrinsic manner (Colombani, 2003).
In multicellular organisms, humoral controls are believed to buffer variations in nutrient levels. However, little is known about how growth of individual cells is coordinated. In vertebrates, growth-promoting action of the growth hormone (GH) is mostly relayed to peripheral tissues through the production of IGF-I. Binding of IGF-I to its cognate receptor tyrosine kinase (IGF-IR) induces phosphorylation of insulin receptor substrates (IRS), which in turn activate a cascade of downstream effectors. These include phospho-inositide 3-kinase (PI3K), which generates the second messenger phosphatidylinositol-3,4,5-P3 (PIP3), and thereby activates the AKT/PKB kinase. Genetic manipulation of IGF-I, IGF-IR, PI3K, and AKT in mice modulates tissue growth in vivo thus demonstrating a requirement of the IGF pathway for growth. In Drosophila, both loss- and gain-of function studies have also exemplified the role of a conserved insulin/IGF signaling pathway in the control of growth. Ligands for the unique insulin receptor (Inr) constitute a family of seven peptides related to insulin, the Drosophila insulin-like peptides (Dilps). Remarkably, three dilp genes (dilp2, dilp3, and dilp5) are expressed in a cluster of seven median neurosecretory cells (m-NSCs) in the larval brain, suggesting that they have an endocrine function. Indeed, ablation of the seven dilp-expressing mNSCs in larvae induces a systemic growth defect (Colombani, 2003).
Both in flies and mice, mutations in IRS provoke growth retardation as well as female sterility similar to what is observed in starved animals. Moreover, PI3K activity in Drosophila larvae depends on the availability of proteins in the food. Overall, this supports the notion that the insulin/IGF pathway might coordinate tissue growth with nutritional conditions. However, upon amino acid withdrawal, neither PI3K nor AKT/PKB activities are downregulated in mammalian or insect cells in culture, suggesting that this pathway does not directly respond to nutrient shortage. Hence, an intermediate sensor mechanism must link nutrient availability to insulin/IGF signaling (Colombani, 2003).
An intriguing possibility is that specific organs could function as nutrient sensors and induce a nonautonomous modulation of insulin/IGF growth signaling in response to changes in nutrient levels. This study used a genetic approach in Drosophila to assess both the cellular and humoral responses to amino acid deprivation in the context of a developing organism. The insect fat body (FB) has important storage and humoral functions associated with nutrition, comparable to vertebrate liver and adipose tissue. During larval stages, the FB accumulates large stores of proteins, lipids, and carbohydrates, which are normally degraded by autophagy during metamorphosis in order to supply the developing tissues but can also be remobilized during larval life to compensate transitory nutrient shortage. In addition to its storage function, the FB also has endocrine activity and supports growth of imaginal disc explants and DNA replication of larval brains in coculture. This study demonstrates that the FB operates as a sensor for variations in nutrient levels and coordinates growth of peripheral tissues accordingly via a humoral mechanism (Colombani, 2003).
In the course of a P[UAS]-based overexpression screen for growth modifiers, a P[UAS]-insertion line (UY681) was found to cause growth retardation upon ectopic activation. Sequence analysis revealed that P(UY)681 is inserted in a predicted gene (CG11128) that encodes a putative protein showing strong homology with amino acid permeases of the cationic amino acid transporter (CAT) family. The P[UAS] element is inserted in the first intron of the CG11128 gene, potentially driving transcription of an antisense RNA in a GAL4-dependent manner. To assess the function of this transporter, 3H-arginine uptake was measured in S2 cells. Results indicate that amino acid uptake is either enhanced by transfection of a CG11128 cDNA or suppressed by RNAi, indicating that the encoded protein presents CAT activity. In situ hybridization revealed basal levels of CG11128 expression in most larval tissues but much higher levels in the FB and the gut, two tissues involved in amino acid processing (Colombani, 2003).
By P element remobilization, an imprecise excision was obtained that deletes the sequences encoding the N-terminal half of the protein. 87% of homozygous mutant animals die during larval stages. The few viable adults emerged after a 2 day delay and were smaller and markedly slimmer than control animals. The associated gene was named slimfast (slif) and the excision allele slif1. Weight measurement indicated that homozygous slif1 adult males displayed a 16% mass reduction compared to control. Accordingly, adult wing size was reduced by 8% due to a reduction of both cell size and cell number. When the slif1 allele was in trans to Df(3L)Δ1AK, a deficiency covering the locus, larval lethality was slightly enhanced, suggesting that slif1 corresponds to a strong hypomorphic allele. The amino acid transporter function of slif, as well as the phenotypes observed upon reduction of slif function suggest that slif mutant animals might suffer amino acid deprivation. A major consequence of amino acid deprivation in larvae is the remobilization of nutrient stores in the FB, which typically results in aggregation of storage vesicles. Consistently, fusion of storage vesicles was observed in the FB of slif1 larvae and was indistinguishable from that observed in animals fed on protein-free media (Colombani, 2003).
GAL4 induction of P(UY)681 resulted in a growth-deficient phenotype similar to that of slif1 loss of function. The antisense orientation of P(UY)681 suggested that the growth defect following GAL4 induction was due to an RNAi effect. Indeed, Northern blot analysis revealed that ubiquitous GAL4-dependent activation of P(UY)681 using the daughterless-GAL4 (da-GAL4) driver strongly reduced slif mRNA levels. Only two of the three alternative first exons are potentially affected by the antisense RNA, possibly explaining the residual accumulation of slif mRNAs in da-GAL4; P(UY)681 animals. Most of these animals died at larval stage, similar to what was observed for slif1 mutants. Specific induction of P(UY)681in the wing disc using the MS1096-GAL4 driver provoked a reduction of the adult wing size, which could be either rescued by coactivation of a UAS-slif transgene or enhanced by reducing slif gene dosage with the heterozygous Df(3L)Δ1AK deficiency. Thus, GAL4-dependent activation of P(UY)681 reduces slif function and defines a conditional loss-of-function allele hereafter termed slifAnti (Colombani, 2003).
As expected, loss of slif function using the slifAnti allele also mimicked amino acid deprivation. Accordingly, ubiquitous slifAnti induction in growing larvae resulted in storage vesicle aggregation and strong reduction of global S6 kinase activity, similar to what was reported in animals raised on protein-free diet. Additionally, an increase in PEPCK1 gene transcription was observed, similar to the effect of amino acid withdrawal. In summary, this study has identified two loss-of-function alleles of the slif gene whose defects mimic physiological aspects of amino acid deprivation. Importantly, the conditional slifAnti allele provides a unique tool to mimic an amino acid deprivation in a tissue-specific manner (Colombani, 2003).
This study established that the FB is a sensor tissue for amino acid levels, as downregulation of the Slif amino acid transporter within the FB is sufficient to induce a general reduction in the rate of larval growth. In contrast, specific disruption of slif in imaginal discs, larval gut, or salivary glands did not induce a nonautonomous growth response, suggesting that these tissues do not participate in the systemic control of growth. The dilp-expressing median neurosecretory cells (m-NSCs) also affect growth control, since selective ablation of these cells in the larval brain induces an overall reduction of animal size. In response to complete sugar and protein starvation, the m-NSCs stop expressing dilp3 and dilp5 genes, suggesting that these neurons also sense nutrient levels. This study shows that the selective reduction of slif function in these cells has no obvious effect on tissue growth and animal development. This indicates that the seven dilp-expressing m-NSCs do not constitute a general amino acid sensor. In contrast, the role of m-NSCs in carbohydrate homeostasis and the observation that they stop expressing certain dilp genes when larvae are deprived of sugar rather suggests that these cells have a role in sensing carbohydrate levels (Colombani, 2003 and references therein).
This analysis also provides a framework in which to understand the phenotype of minidisc, a mutation in an amino acid transporter gene that exhibits nonautonomous growth defects in imaginal discs (Colombani, 2003).
In a number of model systems, both PI3K and TOR have been implicated in linking growth to nutritional status and, until recently, were considered as intermediates of a common regulatory pathway. In yeast, the TOR kinase is part of a cell-autonomous nutrient sensor, which controls protein synthesis, ribosome biogenesis, nutrient import, and autophagy. Genetic analysis in Drosophila indicates that dTOR is required for cell-intrinsic growth control. The results obtained using the slifAnti allele in the wing disc indicate that individual tissues have indeed the potential to respond to amino acid deprivation in a cell-autonomous manner. Nonetheless, this study also demonstrates that the TOR nutritional checkpoint participates in a systemic control of larval growth emanating from the FB. Within a developing organism, each cell may integrate these two distinct inputs regarding nutritional status, one originating from a systemically-acting FB sensor, and the other from TOR-dependent signaling in individual cells. One can further speculate that depending on the strength and duration of starvation, different in vivo nutritional checkpoints will be hierarchically recruited to protect the animal and that the systemic control might, in most physiological situations, override the cell-autonomous control. Indeed, as the data demonstrate, the FB sensor is sufficient to induce a general and coordinated response to starvation without calling individual cell-autonomous mechanisms into play (Colombani, 2003).
Several lines of evidence indicate that the PI3K pathway is not part of the sensor mechanism in FB cells. First, a sensor for PI3K activity in the FB is only marginally affected by amino acid deprivation in that tissue, indicating that the cell-autonomous response to amino acid starvation does not directly influence PI3K signaling. This is reminiscent of previous observations in mammalian cultured cells, showing that PI3K activity does not respond to variations in amino acid levels. Moreover, inhibition of PI3K signaling by dPTEN expression in the FB is not sufficient to trigger the sensing mechanism. Although, dPTEN overexpression causes a complete disappearance of the PI3K sensor accompanied by growth suppression of FB cells, the FB maintains a critical mass that allows for normal larval growth. In contrast, the regulatory subunit p60 whose overexpression potently inhibits PI3-kinase in flies has been shown to induce a systemic effect on larval growth when overexpressed in the FB using an Adh-Gal4 driver. This study found that a pumpless ppl-GAL4-directed expression of p60 also provokes a strong suppression of larval growth and a dramatic inhibition of FB development in young larvae. Thus, the systemic effect on growth observed upon p60 overexpression most likely results from a drastic reduction of FB mass, which then fails to support normal larval growth (Colombani, 2003).
These results further indicate that PI3K signaling is a remote target of the humoral message that originates from the FB in response to amino acid deprivation. This is in agreement with previous data showing that PI3K activity is downregulated by dietary amino acid deprivation and explains why global PI3-kinase inhibition mimics cellular and organismal effects of starvation. The existence of a humoral relay reconciles these in vivo studies with the absence of direct PI3K responsiveness to amino acid levels (Colombani, 2003).
The relative resistance of imaginal disc growth to the systemic control exerted by the FB correlates with maintenance of PI3K activity in these tissues. This is in agreement with previous observations that cells in the larval brain and in imaginal discs maintain a slow rate of proliferation under protein starvation, while larval endoreduplicating tissues (ERTs) arrest. This difference might be attributed to the basal levels of dilp2 expression observed in imaginal discs, allowing a moderate growth rate of these tissues through an autocrine/paracrine mechanism. It was recently shown that clonal induction of PI3K potently induces cell-autonomous growth response even in fasting larvae, indicating that some nutrients are still accessible to support cell growth within a fasted larva. The main function of a general sensor could be to preserve these limited nutrients for use by high priority tissues. In this context, local PI3K activation through an autocrine loop in imaginal tissues could favor the growth of prospective adult structures in adverse food conditions. Thus, the FB would have an active role in controlling the allocation of resources depending on nutritional status. In this respect, it is noteworthy that FB cells are relatively resistant to the FB-derived humoral signal, since the PI3K sensor is not drastically affected in the FB of ppl>slifAnti animals. Thereby, essential regulatory functions of the FB could be preserved even in severely restricted nutritional conditions (Colombani, 2003).
How does the FB signal to other tissues? This study suggests that a humoral signal relays information from the FB amino acid sensor and systemically inhibits PI3K signaling. In addition, this downregulation is not due to a direct inhibition of dilp expression by neurosecretory cells in the brain. Nevertheless, it cannot be ruled out that the secretion of these molecules is subjected to regulation in the mNSCs. Both in vivo and in insect cell culture, several imaginal discs growth factors (IDGF) secreted by the FB have been proposed to function synergistically with Dilp signaling to promote growth. However, this study did not find any modification of IDGF expression in the FB of larvae raised on water- or sugar-only diet, or upon FB induction of slifanti. In vertebrates, the different functions of the circulating IGF-I are modulated through its association with IGF-BPs and acid labile subunit (ALS). In particular, the formation of a ternary complex with ALS leads to a considerable extension of IGF-I half-life. The finding that a Drosophila ALS ortholog is expressed within the FB in an amino acid-dependent manner provides a new avenue to study the molecular mechanisms of nonautonomous growth control mediated by the FB (Colombani, 2003).
This study highlights the contribution that genetics can provide to unravel the mechanisms of physiological control. Using a genetic tool to mimic amino acid deprivation, it was demonstrated that nutrition systemically controls body size through an amino acid sensor operating in the FB. It is proposed that (1) in metazoans, a systemic nutritional sensor modulates the conserved TOR-signaling pathway, and (2) the response to sensor activation is relayed by a hormonal mechanism, which triggers an Inr/PI3K-dependent response in peripheral tissues (Colombani, 2003).
In many species, reducing nutrient intake without causing malnutrition extends lifespan. Like DR (dietary restriction), modulation of genes in the insulin-signaling pathway, known to alter nutrient sensing, has been shown to extend lifespan in various species. In Drosophila, the target of rapamycin (TOR) and the insulin pathways have emerged as major regulators of growth and size. Hence, the role of TOR pathway genes in regulating lifespan has been examined by using Drosophila. Inhibition of TOR signaling pathway by alteration of the expression of genes in this nutrient-sensing pathway, which is conserved from yeast to human, extends lifespan in a manner that may overlap with known effects of dietary restriction on longevity. In Drosophila, TSC1 and TSC2/Gigas (tuberous sclerosis complex genes 1 and 2) act together to inhibit TOR (target of rapamycin), which mediates a signaling pathway that couples amino acid availability to S6 kinase, translation initiation, and growth. Overexpression of dTsc1, dTsc2, or dominant-negative forms of dTOR or dS6K all cause lifespan extension. Modulation of expression in the fat is sufficient for the lifespan-extension effects. The lifespan extensions are dependent on nutritional condition, suggesting a possible link between the TOR pathway and dietary restriction (Kapahi, 2004).
The Drosophila homologs of human Tsc1 (Hamartin) and Tsc2 (tuberin) function in vivo as a complex that controls growth and size in a cell-autonomous manner. To examine their role in regulating lifespan, dTsc1 and dTsc2 were overexpressed through the ubiquitously expressed driver, daughterless (da-GAL4). Overexpression in transgenic flies carrying UAS constructs containing dTsc1 or dTsc2 extends mean lifespan at 29°C by 14% and 12%, respectively. Since GAL4 enhancer traps generally yield stronger effects at 29°C, most of the experiments were performed at that temperature (Kapahi, 2004).
dTsc1 and dTsc2 physically interact with dTOR, which is conserved from yeast to human as a nutrient sensor. Loss of dTsc1 in Drosophila eye leads to an increase in cell size, provided that dTOR is present. Surprisingly, however, dTOR overexpression causes a reduction in cell size, a phenotype similar to dTOR loss-of-function mutations, perhaps due to titration of cofactors required for TOR signaling. The effect of dTOR on lifespan was examined by using three UAS. One carries the full-length wild-type TOR gene. The second carries FRB, the 11 kDa FKBP12-rapamycin binding domain, which has been shown to prevent S phase entry when injected into human osteosarcoma cells. The third carries TED (toxic effector domain), containing the 754 amino acid central region, which inhibits cell growth and arrests cells in G1 when overexpressed in yeast (Hennig, 2002). Ubiquitous overexpression with the da-GAL4 driver of UAS-dTORFRB led to a mean lifespan increase at 29°C of 24%. However, overexpression of UAS -dTORWT or UAS-dTORTED prevented eclosion to adulthood (Kapahi, 2004).
S6 kinase activation upon phosphorylation has been implicated in mediating the downstream effects of TOR on translation initiation in flies and mammals. S6 kinase phosphorylation of ribosomal protein S6 is accompanied by upregulation of a class of mRNAs containing an oligopyrimidine tract at their transcriptional start site termed 5'TOP (Thomas, 2002). Some 200 genes, most of which encode components of the translational apparatus including ribosomal proteins and elongation factors, have this sequence and can account for about 20% of total cellular mRNA. Flies carrying homozygous mutations in dS6K show a developmental delay and a reduction in body size. The stimulation of dS6K phosphorylation by dTOR is abrogated when dTsc1 and dTsc2 are overexpressed. Furthermore, flies with reduced dTSC1 show increased dS6 kinase activation, and genetic reduction of S6 kinase level can rescue the lethality caused by loss of function of dTsc1 (Kapahi, 2004).
The role of S6 kinase in regulating lifespan was examined by using dominant-negative and constitutively active constructs. The dominant-negative effect was achieved by replacing the conserved lysine in the ATP binding site by glutamine (UAS-dS6KKQ), which causes cell-size reduction. The constitutively active form was generated by replacing the phosphorylation sites of S6 kinase by acidic amino acids (UAS-dS6KSTDETE), causing an autonomous cell size increase. By using da-GAL4 to drive ubiquitous overexpression of the dominant-negative form, a mean lifespan increase of 22% at 29°C was observed. Conversely, overexpression of the constutively active form of S6 kinase caused a mean lifespan decrease of 34% at 29°C. Overexpression of dTsc2 and dTORFRB was also tested at 25°C and led to a 20% and 26% increase in mean lifespan increase, respectively (Kapahi, 2004).
To determine which tissues are responsible for the lifespan extension, various GAL4 drivers with specific GAL4 expression pattern were employed to overexpress dTsc2 via a UAS promoter. Overexpression in the eye by using the driver gmr-GAL4 or in the nervous system by using appl-GAL4 did not extend lifespan. In contrast, by using the drivers 24B-GAL4 and PO188-GAL4, enhancer traps that are predominantly expressed in the muscle and fat, results in mean lifespan extensions of 27% and 37%, respectively, at 29°C. The fat-specific drivers DJ634-GAL4 and PO163-GAL4, when used to overexpress dTsc2, also led to a mean lifespan extension of 22% and 31%, respectively, at 29°C. Using DJ634-GAL4 to overexpress the dominant-negative form of TOR (UAS-dTORFRB) or of S6 kinase (UAS-UAS-dS6KKQ) also led to mean lifespan increases of 30% and 29%, respectively, at 29°C. These results indicate that manipulation of the TSC, TOR, and S6 kinase genes in the fat tissue is sufficient for their lifespan extension effects in Drosophila (Kapahi, 2004).
Amino acids have been shown to activate dS6k via TOR, an effect that can be abrogated in the presence of increased levels of dTsc1 and dTsc2. Since nutrients in the diet can modulate lifespan and because the TOR pathway is a critical mediator of nutrient signaling, it was asked whether the observed lifespan-extension effects are dependent on nutrient conditions. This was tested with overexpression of dTsc2 by using the ubiquitously expressing da-GAL4 driver. Flies were allowed to develop to adulthood under standard laboratory food and then maintained on specially prepared food containing various concentrations of yeast extract. At high concentrations of yeast extract, which may be regarded as the opposite of dietary restriction, the lifespan of control flies (da-GAL4/+) is severely reduced. However, overexpression of dTsc2 protects the fly from the deleterious effects of rich food, as if mimicking the effect of dietary restriction. Similar results were observed by overexpression of the dominant-negative form of S6 kinase (Kapahi, 2004).
Recent evidence from Drosophila suggests that signaling through TSC is both parallel to and interacting with the insulin pathway. This is supported by the finding that heterozygosity of dTsc1 or dTsc2 is sufficient to rescue the lethality of loss-of-function dInR mutants. However, the finding that loss-of-function mutations of dTsc1 and dPTEN, a phosphatase that negatively regulates the insulin-signaling pathway, cause cell autonomous and additive increases in cell size suggests that they may be in parallel pathways. Furthermore, in Drosophila, dPTEN loss of function, which leads to an increase in cell size, is only slightly suppressible by loss of function of dFOXO, a fly homolog of C. elegans daf-16. However, the increase in cell size resulting from dTsc1 is enhanced by dFOXO loss of function. Interestingly, unlike long-lived daf-2 mutants, the lifespan extension due to TOR deficiency in C. elegans is not suppressible by a daf-16 mutation. However, the TOR mutant animals do not further extend lifespan in a daf-2 background, leading to the possibility that TOR may be acting downstream or separately from daf-16 to exert its lifespan effects (Kapahi, 2004).
Lifespan extension has been linked with other phenotypes, including stress resistance, metabolic rate, lipid level, reproductive capacity, and body size. The long-lived strains described above with their respective controls for resistance to starvation were compared but no significant differences were found. Similarly, no significant differences were observed for weight and lipid content among these strains. It may be that lifespan extension can be produced by mild modulation of these genes, whereas effects on other phenotypes require severe perturbations. While lifespan extension is observed by using the da-GAL4 driver to overexpress dTsc1 or dTsc2 alone, simultaneous overexpression of dTsc1 and dTsc2 prevented eclosion to adulthood. Similarly, no change in size is observed if dTsc1 or dTsc2 alone are overexpressed in the eye, but a cell-autonomous decrease in size is seen when both are overexpressed simultaneously. Lifespan extension by chico is semidominant, but its effect on body size is recessive. Dominant effects on lifespan are observed with the genes Inr, EcR, Indy, and Rpd3, but their effects on lifespan can be uncoupled from other phenotypes such as fecundity, stress resistance, or lipid accumulation (Kapahi, 2004).
In humans, mutations in TSC1 and TSC2 lead to tuberous sclerosis, a common disorder characterized by the presence of benign tumors in various tissues, with some having large cells. DR in mice has been shown to protect against age-related tumorigenesis. These results suggest a link between lifespan extension by DR and the activities of genes in the TOR pathway. Hence, it is conceivable that the protective effects of DR on tumorigenesis and age-related decline might come from inhibition of such nutrient-responsive pathways (Kapahi, 2004).
These results show that upregulation of dTsc2 in the fat is sufficient for lifespan extension effects in Drosophila. Reduction of daf-2 levels in the C. elegans nervous system has been shown to be sufficient for lifespan extension. However, the lifespan extensions due to mutations in the insulin pathway or germline ablation in C. elegans are dependent on daf-16 activity in the intestine, the fat storage tissue in C. elegans. In Drosophila, the fat body has been proposed to modulate insulin signaling in peripheral tissues by secretion of dALS (acid-labile subunit), which, in mammals, forms a ternary complex with insulin-like growth factor, leading to an extension of the half-life of its ligand. Recently, mice with FIRKO (fat-specific insulin receptor knockout) have been shown to live 18% longer than controls . Hence, it is possible that secondary endocrine signals downstream of the insulin and TOR signaling pathways are released from the fat, and these affect the rate of aging in other tissues. Juvenile hormone and ecdysone are two such endocrine signals that have been implicated in regulating lifespan in conjunction with the insulin pathway in Drosophila (Kapahi, 2004).
Deregulation of Akt/protein kinase B (PKB) is implicated in the pathogenesis of cancer and diabetes. Akt/PKB activation requires the phosphorylation of Thr308 in the activation loop by the phosphoinositide-dependent kinase 1 (PDK1) and Ser473 within the carboxyl-terminal hydrophobic motif by an unknown kinase. This study shows in Drosophila and human cells the target of rapamycin (TOR) kinase and its associated protein rictor are necessary for Ser473 phosphorylation and that a reduction in rictor or mammalian TOR (mTOR) expression inhibits an Akt/PKB effector. The rictor-mTOR complex directly phosphorylated Akt/PKB on Ser473 in vitro and facilitated Thr308 phosphorylation by PDK1. Rictor-mTOR may serve as a drug target in tumors that have lost the expression of PTEN, a tumor suppressor that opposes Akt/PKB activation (Sarbassov, 2005a).
The Akt/PKB kinase is a well-characterized effector of phosphoinositide 3-kinase (PI3K), and its deregulation plays important roles in the pathogenesis of human cancers. PI3K is necessary for the activation of Akt/PKB, and current models suggest that phosphatidylinositol-3,4,5-triphosphates produced upon growth factor stimulation recruit Akt/PKB to the plasma membrane by binding to its N-terminal pleckstrin homology (PH) domain. At the membrane, Akt/PKB is phosphorylated on two key residues: Thr308 (T308) of the activation loop by PDK1 and Ser473 (S473) in the hydrophobic motif of the C-terminal tail by a kinase whose identity has been elusive. The role of S473 phosphorylation is controversial, but there is an emerging view that it precedes the phosphorylation of T308 and is important for the recognition and activation of Akt/PKB by PDK1 (Sarbassov, 2005a and references therein).
The molecular identity of the S473 kinase (S473K), at times referred to as 'PDK2' or the 'hydrophobic motif (HM) kinase,' has been hotly debated for many years. Several candidate S473Ks have been proposed, including PDK1, integrin-linked kinase (ILK), Akt/PKB itself, and, most recently, DNA-PKcs. Many lines of evidence argue that neither PDK1, ILK, nor Akt/PKB is the physiological S473K, and for several reasons, DNA-PKcs is also unlikely to have this function. There is no Drosophila ortholog of DNA-PKcs, and, thus, if DNA-PKcs is a physiological S473K in mammals, a distinct kinase must play that role in flies even though all other core components of the pathway (e.g., PI3K, Akt/PKB, PDK1, and PTEN) are well conserved. Moreover, it has not been shown that DNA-PKcs phosphorylates full-length Akt/PKB, and DNA-PKcs null mice do not suffer the growth retardation or insulin signaling defects associated with Akt1/PKB1 or Akt2/PKB2 (Sarbassov, 2005a).
Mammalian TOR (mTOR) is a large protein kinase that exists in two distinct complexes within cells: one that contains mTOR, GβL, and raptor and another containing mTOR, GβL, and rictor. The raptor-containing complex is sensitive to the drug rapamycin and regulates cell growth, in part by phosphorylating the hydrophobic motif of S6K1, a member of the same family of kinases to which Akt/PKB belongs. The rictor-containing complex does not appear to be rapamycin-sensitive, and its cellular function is just beginning to be understood. Despite its structural similarity to S6K1, Akt/PKB phosphorylation is not sensitive to acute rapamycin treatment, and thus mTOR has not previously been considered as the S473K (Sarbassov, 2005a).
This study used RNA interference (RNAi) in cultured Drosophila cells to determine the role of TOR pathway components in the phosphorylation of the hydrophobic motif sites of Drosophila Akt/PKB (dAKT/dPKB) and S6K (dS6K). In mammals and Drosophila, S6K suppresses signaling through the PI3K/Akt pathway so that inhibition of S6K boosts Akt/PKB phosphorylation. Knockdown of dS6K or Drosophila Raptor expression with double-stranded RNAs (dsRNAs) inhibited the phosphorylation and activity of dS6K and increased the phosphorylation of dAkt/dPKB. Despite reducing dS6K phosphorylation to the same extent as did dRaptor dsRNA, the dTOR dsRNA failed to increase dAkt/dPKB phosphorylation and, surprisingly, decreased it by a small amount. The contrasting effects on dAkt/dPKB phosphorylation by the dTOR and dRaptor dsRNAs suggest that dTOR has an unexpected positive role in dAkt/dPKB signaling that is not shared with dRaptor and that dTOR is required for the increase in dAkt/dPKB phosphorylation caused by dS6K inhibition. Consistent with the dRaptor-independent role for dTOR in dAkt/dPKB phosphorylation, a knockdown of dRictor reduced dAkt/dPKB phosphorylation (Sarbassov, 2005a).
Because basal dAkt/dPKB phosphorylation is low in Drosophila Kc167 cells, the roles of dRictor and dTOR were verified in cells in which dAkt/dPKB phosphorylation was enhanced by decreasing the expression of dPTEN, the negative regulator of the PI3K/Akt pathway. Knockdown of dS6K or dRaptor expression in dPTEN-depleted cells further boosted dAkt/dPKB phosphorylation. In contrast, knockdown of dRictor expression almost completely prevented the dramatic increase in dAkt/dPKB phosphorylation caused by a dPTEN knockdown, whereas the knockdown of dTOR expression caused a slightly smaller suppression. Also, dRictor and dTOR were required for the increase in phosphorylation of dAkt/dPKB caused by a knockdown in the expression of dRaptor (Sarbassov, 2005a).
The results in Drosophila cells suggest that dTOR and dRictor have a shared positive role in the phosphorylation of the hydrophobic motif site of dAkt/dPKB. This finding was unexpected, because previously no decrease was observed in the phosphorylation of the hydrophobic motif site of Akt/PKB after reducing mTOR expression in human cells with small interfering RNAs (siRNAs). In retrospect, however, these experiments were undertaken when RNAi-mediated knockdowns of expression in mammalian cells were relatively inefficient. In this study, with the use of a lentiviral short hairpin RNA (shRNA) expression system that robustly suppresses gene expression, results in human cell lines were obtained analogous to those in Drosophila cells. In human HT-29 colon and A549 lung cancer cells, knockdown of rictor or mTOR expression using two different sets of shRNAs decreased phosphorylation of both S473 and T308 of Akt/PKB. Mammalian cells may try to compensate for the effects of the rictor and mTOR knockdowns by boosting Akt/PKB expression. The decrease in T308 phosphorylation is consistent with the importance of S473 phosphorylation for T308 phosphorylation and with the fact that the Ser473 --> Asp473 mutant of Akt/PKB is a better substrate than the wild-type protein for T308 phosphorylation by PDK1. Knockdown of raptor expression increased the phosphorylation of both S473 and T308 despite reducing Akt/PKB expression. Knockdown of rictor or mTOR expression also decreased S473 phosphorylation in HeLa and HEK-293T cells, two human cell lines that, like A549 and HT-29 cells, contain wild-type PTEN. In addition, the knockdowns also decreased S473 phosphorylation in the PTEN-null PC-3 prostate cancer cell line, a result reminiscent of that in Drosophila cells with reduced dPTEN expression. Furthermore, the knockdowns decreased S473 phosphorylation in M059J glioblastoma cells that are null for DNA-PKcs, a proposed S473K candidate. Thus, in six distinct human cell lines, rictor and mTOR but not raptor are necessary for the phosphorylation of the hydrophobic motif of Akt/PKB (Sarbassov, 2005a).
Because the rictor and mTOR knockdowns inhibit phosphorylation events critical for Akt/PKB activity, they should affect Akt/PKB-regulated effectors. In HeLa cells, a reduction in the expression of rictor or mTOR but not raptor decreased phosphorylation of AFX (Foxo4a), a forkhead family transcription factor that is a direct substrate of Akt/PKB. Because the raptor-mTOR complex directly phosphorylates the hydrophobic motif site of S6K1, whether rictor-mTOR has an analogous function for Akt/PKB was determined. Rictor-mTOR complexes isolated from HEK-293T or HeLa phosphorylated S473 but not T308 of full-length, wild-type Akt/PKB in vitro. Immunoprecipitates of raptor, the ataxia telagiectasia mutated (ATM) protein, or protein kinase C α (PKCα) did not phosphorylate either site, and Akt/PKB did not autophosphorylate S473. Importantly, the raptor immunoprecipitates also contain mTOR but did not phosphorylate Akt/PKB, suggesting that for mTOR to phosphorylate Akt/PKB, it must be bound to rictor and that raptor cannot substitute. This lack of phosphorylation holds even in the raptor immunoprecipitates isolated from HEK-293T cells that contain as much mTOR as the rictor immunoprecipitates. Consistent with a key role for rictor, mTOR immunoprecipitates prepared from the rictor knockdown cells did not phosphorylate Akt/PKB despite containing a similar amount of mTOR as the controls. To verify that mTOR is the S473K in the rictor immunoprecipitates, immunoprecipitates were prepared from control cells and from two different lines of mTOR knockdown cells. Although rictor levels were equivalent in all the immunoprecipitates, only those prepared from cells expressing mTOR phosphorylated Akt/PKB in vitro. Both the LY294002 and wortmannin mTOR kinase inhibitors blocked the in vitro phosphorylation of Akt/PKB by rictor-mTOR, and LY294002 acted at concentrations that inhibit S473 phosphorylation in cells. Staurosporine, an inhibitor of Akt/PKB kinase activity, did not decrease the phosphorylation of Akt/PKB by rictor-mTOR. Thus, in vitro the rictor-mTOR complex phosphorylates S473 of Akt/PKB in a rictor- and mTOR-dependent fashion and with a drug sensitivity profile consistent with mTOR being the phosphorylating kinase (Sarbassov, 2005a).
To determine whether the phosphorylation of Akt/PKB on S473 by rictor-mTOR activates Akt/PKB activity, rictor-mTOR was used to phosphorylate Akt/PKB on S473, and then PDK1 was added to the assay to phosphorylate T308. Prior phosphorylation of Akt/PKB on S473 boosted subsequent phosphorylation by PDK1 of T308, consistent with the importance of S473 phosphorylation for T308 phosphorylation and with the inhibitory effects of the rictor and mTOR knockdowns on T308 phosphorylation. After phosphorylation with rictor-mTOR and PDK1, Akt1/PKB1 had about four- to fivefold more activity than after phosphorylation with PDK1 alone, confirming the important role of S473 in fully activating Akt/PKB. Because growth factors control the phosphorylation of Akt/PKB on S473, it was determined whether the concentration of serum in the cell media regulated the in vitro kinase activity of rictor-mTOR toward Akt/PKB. Rictor-mTOR had decreased activity in HeLa cells deprived of serum and was reactivated by serum stimulation for 30 min, indicating that modulation of the intrinsic kinase activity of rictor-mTOR may be a mechanism for regulating S473 phosphorylation (Sarbassov, 2005a).
These results indicate that the rictor-mTOR complex is a hydrophobic motif kinase for Akt/PKB. Rictor-TOR has essential roles in Akt/PKB hydrophobic motif site phosphorylation in Drosophila and human cells and in vitro phosphorylates full-length, wild-type Akt/PKB in a serum-sensitive fashion. No other proposed hydrophobic motif kinase has been shown to fulfill all these criteria. With hindsight, clues are seen in the literature to the important role of mTOR in Akt/PKB activation. Prolonged but not acute treatment of certain human cells with rapamycin partially inhibits Akt/PKB phosphorylation, and the current findings provide a possible rationale to explain these results. Although rapamycin does not bind to a preformed rictor-mTOR complex, during long-term rapamycin treatment the drug should eventually sequester many of the newly synthesized mTOR molecules within cells. Thus, as the rictor-mTOR complex turns over, rapamycin may interfere with its reassembly or over time become part of the new complexes. It is reasonable to expect then that prolonged rapamycin treatment may partially inhibit rictor-mTOR activity, which would explain why rapamycin is particularly effective at suppressing the proliferation of tumor cells with hyperactive Akt/PKB. The PI3K/Akt pathway is frequently deregulated in human cancers that have lost the expression of the PTEN tumor suppressor gene, and the current findings suggest that direct inhibitors of mTOR-rictor should strongly suppress Akt/PKB activity. Thus, the rictor-mTOR complex, like its raptor-mTOR sibling, may be a valuable drug target (Sarbassov, 2005a).
The insulin/PI3K signaling pathway controls both tissue growth and metabolism. Melted has been identified as a new modulator of this pathway in Drosophila. Melted interacts with both Tsc1 and Foxo and can recruit these
proteins to the cell membrane. Evidence is provided that in the melted mutant, Tor activity is reduced and Foxo is activated. The melted mutant condition
mimics the effects of nutrient deprivation in a normal animal, producing an
animal with 40% less fat than normal (Teleman, 2005).
As a means to identify possible functions of Melted, the
Eukaryotic Linear Motif server) was used
to look for functional motifs
conserved between fly and human Melted. The only conserved motifs found in the
N-terminal region of these proteins were two Forkhead-associated domain ligand
domains (LIG_FHA_1). Forkhead transcription factors FoxA2, FoxA3, FoxC2, and
FoxO1 are involved in glucose and fat metabolism. Insulin signaling activates
Akt, which phosphorylates Foxo and leads to its retention in the cytoplasm. It was
therefore asked if Melted affects the subcellular localization of a Foxo-GFP
fusion protein. Foxo-GFP is predominantly nuclear in the absence of insulin
stimulation in serum-starved S2 cells and increases in the cytoplasm after
insulin stimulation. In
serum-starved cells cotransfected to express Melted, Foxo-GFP is still
primarily nuclear, but much of the
nonnuclear protein appears at the membrane colocalized with Melted. Upon
insulin stimulation, a robust increase in the level of Foxo-GFP was observed at
the cell membrane. The interaction was
confirmed by coimmunoprecipitation of Melted with Foxo in insulin-stimulated S2
cells (Teleman, 2005).
The observation that insulin stimulation
induces a shift toward membrane localization of Foxo in the presence of Melted
in S2 cells raised the possibility that melted regulates Foxo activity in
vivo. To address this, expression of the Foxo target 4E-BP was examined
in wild-type and
melted mutant animals. Under fed conditions, insulin signaling is active
and 4E-BP transcript levels are relatively low.
In wild-type flies that were starved for 24 hr to reduce insulin levels and
thereby activate Foxo, 4E-BP transcript increased ~4-fold. In starved flies
lacking Melted, 4E-BP transcript increased over 25-fold. This increase in 4EBP
transcription was absent in the starved melted/Foxo double mutant, confirming
that it is Foxo dependent. Thus, in the absence of Melted, Foxo activity is
higher than normal, suggesting that Melted limits Foxo activity in vivo (Teleman, 2005).
To determine whether
the elevated Foxo activity observed in melted mutants contributes to the
lean phenotype of these animals, the normalized triglyceride levels
of melted mutant and melted foxo double-mutant flies were compared.
Reducing Foxo activity suppresses the leanness of the melted mutant to a
considerable degree, reaching near normal fat levels.
The rescue was highly statistically significant.
foxo mutants did not show higher-than-normal fat
levels compared to wild-type. These observations suggest that Melted
acts by regulating Foxo activity to control expression of genes important in fat
metabolism (Teleman, 2005).
The Tor pathway integrates information on cellular
nutritional status and stress from the heterodimeric Tsc1/2 complex. melted mutants exhibit reduced Tor activity. By recruiting Foxo to
the membrane in an insulin-regulated manner Melted influences expression of
Foxo targets. By reducing Tor activity and at the same time increasing Foxo
activity, the melted mutant mimics the effects of nutrient deprivation in
a normal animal, producing a lean phenotype (Teleman, 2005).
To determine whether Tor activity affects fat accumulation, the effects were tested
of increasing Tor activity in wild-type and melted
mutant adipose tissue. Use was made of a UAS-Tor transgene that can provide Tor
activity in vivo when expressed at appropriate levels. It was
confirmed that expression of UAS-Tor under ppl-Gal4 control in adipose tissue leads to increased total body fat, as does increasing PI3K
activity. In contrast, a comparable elevation of Tor expression in
melted mutant flies has no effect on fat levels.
Both this result and the significant rescue caused by removal of Foxo
indicate that in the melted mutant, the Foxo branch of the pathway
becomes limiting for fat accumulation. In view of this finding, it was next asked
whether elevated Tor pathway activity could increase fat levels in the
melted mutant if Foxo activity was simultaneously reduced. To do so, use was made of the catalytic subunit of PI3K (Dp110) to inactivate Foxo and
simultaneously activate Tor. The fat body driver lsp2-Gal4 or the UAS-Dp110
transgenes have little effect on their own in the melted mutant
background, but when combined, the elevated PI3K activity in the fat body
increases fat levels of the melted mutant.
The effect is stronger than that of removing Foxo only, increasing fat
levels to above normal. Taken
together, these observations suggest that the Tor branch of the pathway
contributes to the control of fat levels under conditions in which Foxo activity
levels are low. This is normally the case in feeding animals in which insulin
levels are relatively high (Foxo activity is elevated under starvation
conditions: as seen by comparing 4E-BP levels in fed versus starved wild-type and foxo mutant flies). Under conditions in which insulin
levels are low or in the melted mutant, in which Foxo activity is elevated, the effects of Foxo appear to dominate (Teleman, 2005).
The TOR (target of rapamycin) ser/thr protein kinase is the central component of a eukaryotic signaling pathway that regulates growth and is the direct target of the clinically useful drug rapamycin. Recent efforts have identified at least two multiprotein complexes that contain TOR, but little is known in higher eukaryotes about the genes downstream of TOR that control growth. By combining the use of a small molecule inhibitor (rapamycin), transcriptional profiling, and RNA interference in Drosophila tissue culture cells, genes have been identified whose expression responds to Drosophila TOR (dTOR) inhibition and that regulate cell size. Several of the dTOR-regulated genes that function in cell size control have additional roles in cell division. Most of these genes are conserved in mammals and several are linked to human disease. This set of genes is highly enriched for regulators of ribosome biogenesis, which emphasizes the importance of TOR-dependent transcription in building the protein synthesis machinery in higher eukaryotes. In addition, two dTOR-regulated genes, CG3071 and CG6677, have been identified whose human orthologs, SAW and ASH2L, are also under TOR-dependent transcriptional control and encode proteins with conserved functional roles in growth. It is concluded that combining RNA interference with genomic analysis approaches, such as transcriptional profiling, is an effective way to identify genes functioning in a particular biological process. Moreover, this strategy, if applied in model systems with simpler genomes, can identify genes with conserved functions in mammals (Guertin, 2006).
This study defines growth as an increase in cell mass, which is distinct from cell division, although the two processes are coordinated. How coordination is achieved is still debated, but in many cells, cell-cycle progression requires growth. From this model has emerged a hypothesis that a 'size threshold' exists that prevents cell-cycle progression until a critical size is attained. When assessing size, it is not clear what cells measure, but the rate of protein synthesis is a good prediction. Therefore, it is not surprising that rapamycin treatment (which is thought to mimic nutrient deprivation) would change the expression of genes with roles in various aspects of setting the protein synthesis rate (Guertin, 2006).
Studies in Saccharomyces cerevisiae investigating the role of TOR in transcription have determined that yeast TOR regulates expression of metabolic pathway genes, and in addition, ribosomal RNA and ribosomal protein genes; transcription, translation, and replication factors; and protein degradation genes. Studies with mammalian cells have indicated that rapamycin affects expression of a diverse array of genes functioning in nutrient and protein metabolism. But in these studies, a functional connection to growth was not established. Ribosomal proteins and ribosome assembly factors have a critical role in controlling growth. Furthermore, two proteins (Sfp1 and Sch9—an AGC kinase related to AKT and S6K) that activate expression of transcriptional units encoding ribosomal proteins and ribosome assembly factors are regulated in part by TOR. This study study suggests that in higher eukaryotes, TOR regulates expression of genes functioning in multiple steps in the ribosome assembly pathway. But importantly, it was found that many rapamycin-responsive genes that are necessary for normal growth encode ribosome biogenesis regulators, a connection not made in higher organisms (Guertin, 2006).
Previous genetic studies in Drosophila were consulted to determine if any of the genes identified in this study by a cell culture-based system have previously been shown to have a role in organismal growth. One positive growth gene that was identified was CG3333/Nop60B. Partial loss of Nop60B (also named minifly) function results in severe reduction in body size and developmental delay (Giordano, 1999). One positive growth and proliferation gene identified, CG6375/pitchoune/pit, is also required for cell growth and proliferation in developing Drosophila larvae (Zaffran, 1998). Finally, CG5786/ppan (peter pan), a gene identified in a screen for larval growth regulators, was also found to be required for normal growth and proliferation of cultured cells (Migeon, 1999). These findings suggest that this approach led to the identification of physiologically relevant genes (Guertin, 2006).
Nutrient control of gene expression and growth has emerged as a principal concern in the modern era as diet-induced diseases become more prevalent. It is believed that mTOR is at the core of an ancient growth pathway that senses nutrient levels, particularly amino acids, and that rapamycin treatment mimics a 'starvation-like' state. A previous study employing Affymetrix microarrays identified a set of genes in developing Drosophila larvae that respond to starvation. 42% of the 19 genes identified by Zinke (2002) as responding negatively to amino acid starvation were also rapamycin-sensitive genes: a remarkable similarity considering the difference in source material and statistical analysis. If all the genes whose expression decreased significantly (p < 0.01) after rapamycin treatment are considered, it was found that 68% of the genes in the list of Zinke (2002) are also in the current list. While none of the genes Zinke identified as significantly increasing expression upon starvation (14 total) passed cutoffs to be functionally analyzed in this study, 4 of them (28%) did significantly increase expression in response to rapamycin. One of those genes encodes Thor, the Drosophila 4E-BP ortholog. Both the Zinke study and the current study used early versions of the Drosophila Affymetrix chip ('DrosGenome1'), and therefore there might be additional nutrient- and rapamycin-sensitive genes to be discovered (Guertin, 2006).
An appreciation for the role of mTOR in tumorigenesis has emerged from clinical trials indicating that rapamycin might be an effective treatment for some cancers. A general idea is that upregulated mTOR signaling provides tumors with a growth advantage by promoting translation initiation through S6K1 and 4E-BP1. However, it cannot be ruled out that other mTOR-regulated process such as transcription and autophagy are relevant in cancer and other diseases. These processes are conserved in yeast and represent ancient functions of TOR. Perhaps mTOR-regulated genes could be important targets for antigrowth drugs. The fact that many of the growth genes identified block the TSC2 RNAi-induced cell size increase when cosilenced supports such a notion (Guertin, 2006).
Genes with specific links to human disease were also identified in this report. Nop60B (CG3333) is the Drosophila ortholog of human DKC1 (dyskerin), the gene mutated in dyskeratosis congenita (DC). DC is a rare X-linked recessive disease initially characterized by nail dystrophy, abnormal skin pigmentation, mucosal leucoplakia, and premature aging, with patients often succumbing to bone marrow failure before the age of 30. DKC1 encodes a pseudouridine synthase, which associates with box H/ACA small nuclear RNAs and posttranscriptionally modifies rRNA by converting uridine to pseudouridine. In yeast, the DKC1 ortholog (Cbf5p) associates with Nhp2p, the Drosophila ortholog of which (CG5258) was also identified in the screen. Patients with DC are predisposed to tumor formation, and this is mimicked in a mouse model in which half of Dkc1 mutant animals develop tumors. It is perhaps paradoxical that mutations apparently compromising ribosome function promote tumorigenesis. However, a screen for cancer genes in zebrafish identified several ribosomal protein genes as haploinsufficient tumor suppressors, suggesting that ribosome dysfunction may have an important but undefined role in promoting tumor formation (Guertin, 2006).
The human ash2/CG667 gene product (ASH2L) was discovered in a histone methyltransferase (HMT) complex with the tumor suppressor menin (see Drosophila Menin-1). Menin is encoded by the MEN1 gene, which is mutated in familial multiple endocrine neoplasia type 1. Several MEN1 point mutations found in tumors are associated with reduced HMTase activity of the complex. Another report found ASH2L associated with a HMT complex containing the Leukemia protooncoprotein MLL, the human ortholog of Drosophila trithorax, in addition to menin. Interestingly, Drosophila ash2 mutant cells in genetic mosaics exhibit defective cell differentiation and increased cell size, consistent with these conclusions. In another report, Drosophila ASH2 localized to the nucleolus, suggesting that ASH2 might have a role in rDNA transcription. The yeast ortholog of ASH2 is part of the SET1 complex, which is reported to repress rDNA transcription by promoting H3 Lys4 methylation of rDNA. Elucidating the function of ASH2 in cell growth and differentiation might uncover clues to understanding the tumor-suppressor functions of these HMT complexes (Guertin, 2006).
This study combined gene expression profiling with functional analysis by RNAi to identify Drosophila genes that are responsive to acute rapamycin treatment and that regulate cell growth and proliferation. This approach allowed functional annotation of 54 Drosophila genes. Most of the genes have orthologs in species ranging from yeast to mammals, and some are implicated in human disease. With genome-scale RNAi libraries becoming available in many organisms, it is concludes that similar combinatorial approaches might be useful in determining subsets of 'enriched' genes that could be functionally analyzed by targeted RNAi. This study found a rapamycin-sensitive growth-gene set in Drosophila cultured cells. In addition to emphasizing the role of TOR-dependent transcription in growth, human genes were identified with similar transcriptional and functional roles. It is further concluded that similar approaches in model systems with comparatively simpler genomes can be an effective way to predict human gene function (Guertin, 2006).
Target of rapamycin (TOR) is a central regulator of cellular and organismal growth in response to nutrient conditions. In a genetic screen for novel TOR interactors in Drosophila, the clathrin-uncoating ATPase Hsc70-4, which is a key regulator of endocytosis, was identified. Genetic evidence is presented that TOR signaling stimulates bulk endocytic uptake and inhibits the targeted endocytic degradation of the amino acid importer Slimfast. Thus, TOR simultaneously down-regulates aspects of endocytosis that inhibit growth and up-regulates potential growth-promoting functions of endocytosis. In addition, disruption of endocytosis leads to changes in TOR and phosphatidylinositol-3 kinase activity, affecting cell growth, autophagy, and rapamycin sensitivity. These data indicate that endocytosis acts both as an effector function downstream of TOR and as a physiologically relevant regulator of TOR signaling (Hennig, 2006; full text of article).
Inactivation of TOR causes an inhibition of cellular growth, a reduction in cell size, and a suppression of cell cycle progression. In addition to well described changes in protein synthesis and ribosome biogenesis, recent studies have suggested that other cell processes are likely to contribute to these growth effects of TOR. The present study identifies endocytosis as one such process. These results demonstrate that the clathrin-uncoating ATPase Hsc70-4 interacts genetically with TOR and Tsc1, and that bulk endocytosis is stimulated in cells with activated TOR signaling. Conversely, it was found that TOR activity inhibits the endocytic degradation of nutrient transporters such as Slimfast. Together, these endocytic effects of TOR promote both the bulk and targeted uptake of nutrients and other biomolecules required for cell mass increase. In addition to this direct role in cellular biosynthesis and growth, nutrients also act as potent regulators of TOR signaling. Indeed, Slimfast has been identified as an upstream activator of TOR (Colombani, 2003). These findings that disruption of endocytosis effects cell size, rapamycin sensitivity, and TOR kinase activity are consistent with an additional role for endocytosis upstream of TOR (Hennig, 2006).
Mutations that disrupt endocytosis are likely to have both positive and negative effects on nutrient uptake and cell growth because they inhibit bulk endocytic uptake, as well as degradation of nutrient transporters and other signaling molecules. Thus, the overall effects of endocytic disruption on nutrient uptake, cell growth, and TOR signaling are difficult to predict a priori. The results suggest that both the cellular context and the specific step at which endocytosis is blocked influence the growth response. Thus, in fat body cells, expression of ShiK44A resulted in an increase in cell size, whereas loss of Hsc70-4 function caused reduced cell size. It is noted that these changes mirror the effects of these mutations on Slimfast levels; whereas both ShiK44A expression and Hsc70-4 mutation decreased bulk endocytic uptake, only ShiK44A resulted in increased levels of Slimfast. In contrast, both ShiK44A and Hsc70-4 mutants led to the increased size of wing imaginal disc cells, suggesting that in these cells the growth-inhibitory effects of endocytic degradation of membrane proteins such as Slimfast predominate over the potential positive effects of increased bulk uptake. Similarly, the results indicate a complex effect of endocytosis on TOR signaling. Partial reduction in Hsc70-4 levels lead to an increase in TOR signaling, as was evident in an eyTOR interaction and rapamycin resistance. In contrast, larvae that are homozygous mutant for Hsc70-4 show a decrease in TOR kinase activity. These results suggest that modest inhibition of endocytosis may increase TOR signaling, whereas a complete block of endocytosis may reduce it (Hennig, 2006).
A striking parallel to the inverse regulation of bulk and targeted endocytic processes by TOR can be observed in its effects on autophagy in yeast. Through autophagy, random portions of cytoplasm are nonselectively engulfed within double membrane–bound vesicles for delivery to the lysosome. Activation of TOR causes this nonselective form of autophagy to be suppressed, and, instead, the autophagic machinery engages in a selective type of autophagy known as the cytoplasm–vacuole targeting (CVT) pathway, which is responsible for lysosomal delivery of specific hydrolases. Thus, TOR acts as a switch between selective and nonselective autophagy. TOR may also be involved in switching between clathrin-and caveolae/raft-mediated endocytosis in higher eukaryotes. A genome-wide survey of protein kinases found that RNAi-mediated inactivation of TOR in HeLa cells inhibited clathrin-dependent processes such as transferrin uptake and vesicular stomatitis virus infection, and stimulated cavelolae/raft-dependent events (Pelkmans, 2005). Together, these findings suggest that TOR may control the specificity of membrane trafficking components. In addition, the results show that S6K, which is an important TOR substrate, acts downstream of TOR in promoting bulk endocytosis, but is not involved in the suppression of starvation-induced autophagy (Hennig, 2006).
The identification of endocytosis as a TOR-controlled function adds to the growing list of cell processes regulated by TOR, including protein synthesis, ribosome biogenesis, autophagy, metabolic gene expression, and cytoskeletal organization. How these distinct functions interact to achieve a coordinated growth response is only beginning to be understood. One likely mechanism involves the common use of molecular components and cellular substrates by different cell functions, as in the case of selective and nonselective autophagy, bulk endocytosis, and endocytic degradation. Two or more distinct branches of TOR signaling may also act cooperatively to control the same target, as in the case of Slimfast regulation by both translation and endocytosis, or may act in opposition, as previously observed for the role of S6K in limiting autophagy. Finally, distinct TOR complexes may converge on the same targets with opposing effects, as in the regulation of Akt by TOR-raptor versus TOR–rictor complexes (Shah, 2004; Sarbassov, 2005a). The finding that TOR signaling regulates the levels of Slimfast, which was previously shown to function upstream of TOR, adds another layer of complexity to the TOR signaling network (Hennig, 2006).
Previous studies have demonstrated reexpression of cell-cycle markers within postmitotic neurons in neurodegenerative tauopathies, including Alzheimer's disease (AD). However, the critical questions of whether cell-cycle activation is causal or epiphenomenal to tau-induced neurodegeneration and which signaling pathways mediate cell-cycle activation in tauopathy remain unresolved. Cell-cycle activation accompanies wild-type and mutant tau-induced neurodegeneration in Drosophila, and genetically interfering with cell-cycle progression substantially reduces neurodegeneration. The data support a role for cell-cycle activation downstream of tau phosphorylation, directly preceding apoptosis. Accordingly it is shown that ectopic cell-cycle activation leads to apoptosis of postmitotic neurons in vivo. As in AD, TOR (target of rapamycin kinase) activity is increased in this model and is required for neurodegeneration. TOR activation enhances tau-induced neurodegeneration in a cell cycle-dependent manner and, when ectopically activated, drives cell-cycle activation and apoptosis in postmitotic neurons. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model, identifying TOR and the cell cycle as potential therapeutic targets in tauopathies and AD (Khurana, 2006).
It was first determined whether cell-cycle activation accompanied neurodegeneration in a fly model of tauopathy. Expression of a mutant form of tau linked to familial frontotemporal dementia, tauR406W, in the fly brain (panneural driver: ELAV-GAL4) leads to progressive neurodegeneration (Wittmann, 2001). At eclosion, the brains of tau-expressing flies appear morphologically normal, but by 10 days clear neurodegeneration is observed, characterized histologically by condensation and fragmentation of neuronal nuclei and vacuolization. TUNEL staining identified apoptotic neurons in tau transgenic animals but not in age-matched controls (Khurana, 2006).
PCNA and PH3 were immunostained to assess early and late cell-cycle activation. Control animals were completely negative for PCNA and the M-phase marker phosphohistone-3 (PH3) at 10 days and 30 days. In contrast, brains from tau transgenic flies showed prominent expression of both PCNA and PH3 at 10 days. PCNA staining was particularly prominent in areas of neurodegeneration, as indicated by characteristic nuclear changes and cytoplasmic vacuolization. Together, these findings demonstrate that abnormal activation of the cell cycle accompanies tau-induced apoptotic neurodegeneration in Drosophila and suggest that cell-cycle activation is likely to be a relatively late event in this model (Khurana, 2006).
This study established a causal relationship between cell-cycle activation and tau-induced neurodegeneration in vivo. Expression of both mutant and wild-type tau induce cell-cycle activation in this model, and genetic inhibition of the cell cycle substantially reduces tau-induced neurodegeneration in both the fly brain and retina. The issue of causality has previously been unresolved, although important studies have documented aberrant neuronal cell-cycle markers in human tauopathies and, more recently, in a mouse tauopathy model. The data also provide in vivo support for key experiments in cell-culture systems that have demonstrated cell cycle-dependent apoptosis in a variety of neurotoxic paradigms. Previous reports have implicated cell-cycle activation in rodent models of stroke and head trauma, although these studies have largely relied upon pharmacologic inhibition of the cell cycle by Cdk inhibitor drugs. Cdk inhibitors target several non-cell-cycle kinase targets, including GSK-3 and Cdk5, that have also been implicated in cell survival and tau phosphorylation. Indeed, while Cdk inhibitors were recently shown to be neuroprotective in a toxic mouse model of PD, this effect was found to be more attributable to inhibition of Cdk5 than to inhibition of cell cycle-related kinases (Khurana, 2006).
In this study, PCNA-, PH3-, and TUNEL-positive neurons were demonstrated in the tauopathy model and it was concluded that cell-cycle activation and apoptosis accompanied neurodegeneration. While expression of PCNA and PH3 have been described in processes other than cell division (DNA repair and immediate early gene responses, respectively), the genetic data implicating multiple components of the cell cycle in tau-induced neurodegeneration strongly support a cell-cycle role in this model. While TUNEL-positive cell death may not always be apoptotic, previous reports showing that antiapoptotic genes, including IAP-1, block tau-induced neurodegeneration in flies support a role for apoptosis in this model. The role of apoptosis in tauopathies and animal tauopathy models remains controversial, however. Recently, both apoptotic and nonapoptotic neurodegeneration were described in a mouse tauopathy model, and the possibility cannot be ruled out that nonapoptotic forms of cell death occur in the fly model also (Khurana, 2006).
The relationship between cell-cycle activation and tau phosphorylation has previously been unclear. Since Cdks are proline-directed kinases known to phosphorylate tau in vitro, cell-cycle activation could mediate neurodegeneration by directly phosphorylating tau. Indeed, several serine and threonine residues of tau are substrates for Cdks in vitro, and mitosis in cultured proliferating cells is associated with tau phosphorylation at these sites. Second, cell-cycle activation could be downstream of phosphorylation and directly lead to apoptosis in two plausible ways. First, forcing differentiated cells to enter a cell cycle could directly lead to apoptosis via an aborted attempt to replicate damaged DNA. Such a mechanism may be particularly relevant to postmitotic neurons that are known to have a limited capability for DNA repair. Alternatively, it is possible that cell-cycle mediators, including E2F1 and Cdk1, may subserve dual functions as direct mediators of neuronal apoptosis (Khurana, 2006).
The data support a role for cell-cycle activation downstream of tau phosphorylation and directly preceding apoptosis. First, cell-cycle markers often immunolocalized to areas characterized histologically by nuclear fragmentation and condensation, suggesting a late role in neurodegeneration. Second, cell-cycle modulation dramatically modifies tau-induced neurodegeneration without altering tau phosphorylation at disease-associated epitopes that can be generated by Cdks in vitro. In contrast, pseudophosphorylation of tau or reducing tau phosphorylation in a sgg mutant background directly increased and decreased cell-cycle activation, respectively. Third, cell-cycle modulation could still enhance toxicity of tauE14, a pseudophosphorylated construct in which all Ser-Pro and Thr-Pro target sites are mutated to glutamate. Fourth, double labeling of brains of tau-expressing flies for PH3 and tau phosphoepitopes revealed that >90% PH3-positive neurons were phosphoepitope positive, even for relatively restricted epitopes such as AT-180 (20% of all neurons). Finally, it was shown that cell-cycle activation, in the absence of transgenic tau, could directly lead to apoptosis of postmitotic neurons in vivo, supporting the possibility that cell-cycle activation could directly transduce tau-induced apoptosis (Khurana, 2006).
The mechanisms through which cell cycle becomes activated in tauopathies have not been defined. Markers that could represent aberrant mitogenic signaling are aberrantly expressed in these diseases, including markers of MAP kinase activation, classic oncogenic pathways such as Src and c-Myc, and TOR activation. However, the expression of isolated markers, while interesting, establishes neither the importance of a particular pathway as a whole nor whether any of these pathways are able to reactivate cell cycle in postmitotic neurons or lead to neurodegeneration in vivo. In this study, it was shown that TOR activation occurs in the current tauopathy model, recapitulating a similar finding in AD, and is furthermore required for neurodegeneration. Since TOR-dependent enhancement of tau toxicity is blocked by concomitant cell-cycle inhibition, and ectopic TOR activation leads to neuronal cell-cycle activation and apoptosis in the adult fly brain, the data indicate that TOR signaling mediates tau-induced neurodegeneration via cell-cycle activation. The relationship between tau, TOR, and cell-cycle activation, however, may be complex. In the retina, for example, TOR activation in the absence of transgenic tau results in an enlarged eye, whereas tau expression results in a small, rough eye. It is plausible that these phenotypic differences may be related to tau-induced pathogenic events that occur upstream of TOR activation in the described model. Also, while the data strongly link TOR to cell-cycle activation in the model, other TOR-dependent mechanisms of neurotoxicity cannot be ruled out. For example, TOR activation could theoretically enhance neurodegeneration by inhibiting autophagy, although the rescue of tau toxicity by loss of S6k would argue against this possibility since S6k is an activator of autophagy in flies (Khurana, 2006).
Aging is a significant risk factor for tauopathies. Interestingly, TOR inhibition is known to prolong lifespan in Drosophila, and the data thus directly link an aging-related signal transduction pathway to tau-induced neurodegeneration. Furthermore, withdrawal of amino acids in vitro or starvation in vivo results in inhibition of TOR signaling, potentially offering a molecular mechanism for the neuroprotection reported in human studies by caloric restriction (Khurana, 2006).
In this study, the role of cell-cycle activation in tau-mediated neurodegeneration was investigated because aberrant expression of cell-cycle markers is best described for tauopathies and AD. For example, one comprehensive study found upregulation of cell-cycle markers in AD and in a cohort of sporadic and inherited tauopathies but not in other diseases including PD, Huntington's disease, amyotrophic lateral sclerosis (ALS), or multiinfarct dementia. However, others have reported cell-cycle marker upregulation in the context of spinal cord injury, ALS, and PD. In addition, neuronal expression of cell-cycle markers has been described in several cell-culture paradigms of neurotoxicity and in mouse models of ataxia, leading to the speculation that cell-cycle activation might be a universal mechanism for neurodegeneration, perhaps related to oxidative stress. In the current study, however, no evidence of cell-cycle activation was found in fly models of either Machado Joseph Disease (MJD) protein or Parkinson's disease, despite the presence of apoptotic neurodegeneration, and cell-cycle modulation did not modify MJD-induced neurodegeneration. These data implicate distinct mechanisms for neurodegeneration in different neurodegenerative diseases, consistent with recent findings that forward genetic screen modifiers differ between polyglutamine-, synuclein-, and tau-induced neurodegeneration (Khurana, 2006).
In summary, the results indicate a common effector pathway and potentially common therapeutic strategies for cancer and tauopathy, two major causes of age-related morbidity and mortality. The TOR signaling pathway, a known regulator of lifespan, was delineated as a required mediator of tau-dependent neurodegeneration in vivo. The results provide a rationale for the assessment of TOR and cell-cycle inhibitors as potential therapeutic strategies in tauopathies and AD. (Khurana, 2006).
Phosphatidylinositol-3-kinase (PI3K)/AKT signaling is essential for growth and metabolism and is elevated in many cancers. Enzymatic activity of AKT has been shown to depend on phosphorylation of two conserved sites by PDK1 and TOR (target of rapamycin) complex 2 (TORC2) in a PI3K-dependent manner. This study analyzed the role of TORC2-mediated AKT phosphorylation in Drosophila. Mutants removing critical TORC2 components, rictor and sin1, strongly reduced AKT hydrophobic motif (HM) phosphorylation and AKT activity, but showed only minor growth impairment. A mutant form of AKT lacking the HM phosphorylation site displayed comparable activity. In contrast to the mild effects of removing HM site phosphorylation at normal levels of PI3K activity, loss of TORC2 activity strongly inhibited hyperplasia caused by elevated pathway activity, as in mutants of the tumor suppressor PTEN. Thus, TORC2 acts as a rheostat to broaden the range of AKT signaling at the high end of its range (Hietakangas, 2007).
The PI3K/AKT signaling pathway is conserved between Drosophila and mammalian species. The lack of genetic redundancy among pathway components makes Drosophila a useful system in which to dissect the roles of the individual pathway members in vivo. Earlier analyses of other pathway members have shown that the Insulin receptor, PI3K, PDK1, and AKT are each essential for viability, and that mutant tissue displays severe undergrowth. Mutants of Drosophila insulin receptor substrate homolog, chico, are semiviable but severely growth impaired. Although individual AKT mutants are viable in mouse, the essential nature of AKT is likely to be masked by genetic redundancy among the three AKT genes. Previous studies in cultured cells have suggested that TORC2 is an important regulator of AKT phosphorylation and activity, and that this phosphorylation event is required for AKT kinase activity. It was recently shown that loss of TORC2 activity in rictor mutant mice leads to loss of AKT HM phosphorylation and to embryonic lethality, suggesting that HM phosphorylation is essential for AKT activity in the mouse. In contrast, the current findings show that TORC2-mediated phosphorylation on the HM site is not essential for AKT activity in vivo. Indeed, although AKT activity was reduced, considerable residual activity was found in flies lacking TORC2 activity. Flies expressing a mutant form of AKT lacking the HM phosphorylation site also showed considerable AKT activity in vivo. These findings indicate that the maximal level of AKT activity is limited in the absence of HM phosphorylation. Under normal physiological conditions in Drosophila, this reduced level of AKT activity is almost sufficient to support normal growth. But without HM phosphorylation, AKT cannot transduce the higher-than-normal levels of PI3K pathway activity that result from mutation of the tumor suppressor PTEN or increased insulin stimulation. When considered in this context, the lethality of rictor mutant mice could reflect a higher threshold in the requirement for AKT activity in some biological process in mouse than in fly, but the possibility of essential TORC2 targets other than AKT cannot be excluded (Hietakangas, 2007).
Perhaps the most intriguing implication of this study lies in the area of cancer biology. Elevated AKT activity is a hallmark of human cancer, with a substantial proportion of human tumors depending on AKT pathway activation, for example, due to PTEN mutations. The current findings suggest that inhibiting TORC2 activity, rather than AKT itself, may prove to be a promising strategy for cancer therapy (Hietakangas, 2007).
To survive starvation and other forms of stress, eukaryotic cells undergo a lysosomal process of cytoplasmic degradation known as autophagy. Autophagy has been implicated in a number of cellular and developmental processes, including cell growth control and programmed cell death. However, direct evidence of a causal role for autophagy in these processes is lacking, due in part to the pleiotropic effects of signaling molecules such as TOR that regulate autophagy. This study circumvents this difficulty by directly manipulating autophagy rates in Drosophila through the autophagy-specific protein kinase Atg1 (Atg signifies an autophagy-related gene). Overexpression of Atg1 is sufficient to induce high levels of autophagy, the first such demonstration among wild type Atg proteins. In contrast to findings in yeast, induction of autophagy by Atg1 is dependent on its kinase activity. Cells with high levels of Atg1-induced autophagy are rapidly eliminated, demonstrating that autophagy is capable of inducing cell death. However, this cell death is caspase dependent and displays DNA fragmentation, suggesting that autophagy represents an alternative induction of apoptosis, rather than a distinct form of cell death. In addition, this study demonstrates that Atg1-induced autophagy strongly inhibits cell growth, and that Atg1 mutant cells have a relative growth advantage under conditions of reduced TOR signaling. Finally, this study shows that Atg1 expression results in negative feedback on the activity of TOR itself.
These results reveal a central role for Atg1 in mounting a coordinated autophagic response, and demonstrate that autophagy has the capacity to induce cell death. Furthermore, this work identifies autophagy as a critical mechanism by which inhibition of TOR signaling leads to reduced cell growth (Scott, 2007).
Under starvation conditions, eukaryotic cells recover nutrients via autophagy, a lysosome-mediated process of bulk cytoplasmic degradation. Through autophagy, long-lived proteins, organelles, and other components of the cytoplasm are non-selectively engulfed within specialized double-membraned vesicles known as autophagosomes. Subsequent fusion of the outer autophagosomal membrane with the lysosome results in a structure known as the autolysosome, in which the inner membrane and its cytoplasmic cargo are degraded. Breakdown products released from the autolysosome supply the cell with an internal source of nutrients that can support essential metabolic processes during starvation. Approximately 20 ATG (autophagy-related) genes specifically required for autophagy have been discovered in Saccharomyces cerevisiae, and many of these genes have functional homologs in metazoans (Scott, 2007 and references therein).
In addition to survival of starvation, autophagy has been implicated in many aspects of health and development, including aging, programmed cell death, pathogenic infection, stress responses, neurodegenerative and muscle disorders, cellular remodeling, cancer, and cell growth. As in many of these processes, evidence for a role of autophagy in cell growth control is largely correlative. Autophagy is promoted by several tumor suppressor genes including PTEN, TSC1, TSC2, and beclin 1, and is inhibited by growth-promoting pathways such as type I PI3K and target of rapamycin (TOR) signaling. A variety of conditions that stimulate cell growth, such as growth factor addition, partial hepatectomy, and refeeding after starvation, also inhibit autophagy, while growth suppressive signals, such as contact inhibition and substrate detachment, induce autophagy. Together, these correlative findings are consistent with a model in which the catabolic effects of autophagy act as a brake on cell growth during development. However, this issue is complicated by the pleiotropic nature of the signaling pathways that regulate autophagy. For example, in addition to inhibiting autophagy, the TOR pathway controls cell metabolism and biosynthesis by promoting ribosome biogenesis, protein synthesis and nutrient uptake. The relevant contribution of each of these downstream effector pathways to net cell growth is poorly understood (Scott, 2007).
Autophagy is also known to be involved in programmed cell death, and distinguishes Type II (autophagic) from Type I (apoptotic) cell death. Autophagic cell death is characterized by an abundance of autophagosomes and autolysosomes in the dying cell and differs from apoptotic cell death in that dying cells are degraded by their own lysosomal enzymes, rather than by phagocytosis. It has been difficult, however, to establish whether autophagy plays a causal role in Type II cell death or represents a failed attempt at cell survival in cells undergoing programmed cell death. Death signals often induce features of both apoptosis and autophagy, and mutations that disrupt autophagy have been shown to suppress cell death in some cases, and hasten it in others. Thus, as in the case for cell growth, much of the support for a direct role for autophagy in cell death rests on correlative evidence (Scott, 2007).
One approach to addressing the potential role of autophagy in functions such as cell death and growth would be to induce autophagy directly, independent of the signaling pathways that normally control it. In yeast, multiple signaling pathways, including TOR, AMPK and Ras/PKA, converge on the Ser-Thr kinase Atg1 to regulate autophagy. In addition, Atg1 interacts with multiple components of the autophagic machinery, through direct association, phosphorylation, and/or through effects on intracellular localization). Thus, Atg1 may represent a nodal point for controlling multiple steps in the autophagic process in response to various inductive cues (Scott, 2007).
Interestingly, the role of Atg1 kinase activity in yeast is the subject of some debate, and studies from different groups using ATP analog-sensitive and kinase-defective Atg1 mutants have reached conflicting conclusions. One study reported that Atg1 kinase activity is required for the autophagy-related biosynthetic cytoplasm to vacuole targeting (CVT) pathway but not for autophagy, and concluded that Atg1 plays a structural role in autophagy. However, other studies using similar reagents found a requirement for Atg1 kinase activity in both CVT and autophagy (Scott, 2007 and references therein).
It has been shown that the Drosophila homolog of Atg1 is required for autophagy in the larval fat body, an organ analogous to the vertebrate liver with roles in nutrient storage and mobilization. This study investigated the effects of Drosophila Atg1 loss of function and overexpression on autophagy induction, cell growth control, and cell death. The findings indicate that Atg1 expression is sufficient to effect a full autophagic response, resulting in a marked inhibition of cell growth and a rapid induction of apoptotic cell death (Scott, 2007).
Delivery of cytoplasmic components to the lysosome through autophagy involves multiple distinct steps including nucleation, expansion and closure of the autophagosome, and its subsequent fusion with the lysosome. These membrane trafficking events require the recruitment and subsequent retrieval of a large number of autophagy-specific Atg proteins, as well as general factors involved in vesicle trafficking. Given this complexity, the finding that overexpression of a single Atg protein is sufficient to accomplish all essential steps in this process is striking. As autophagy occurs constitutively at a basal rate in most eukaryotic cells, acceleration of a single rate-limiting step by overexpression of Atg1 may be sufficient to increase the overall rate of autophagy. Alternatively, the interaction of Atg1 with multiple Atg proteins suggests that Atg1 may function at multiple steps in the autophagic process. Identification of the relevant in vivo substrates of Atg1 will help to clarify this issue. A recent yeast proteomic microarray study identified a number of in vitro substrates of Atg1, including Atg8 and Atg18, which function in autophagosomal expansion and retrieval of components of the autophagic machinery, respectively, as well as general factors involved in vesicle transport and vacuolar function (Scott, 2007).
The role of the kinase activity of yeast Atg1 in regulating autophagy is the matter of some debate, since different groups have drawn conflicting conclusions. A consensus view, however, may be that autophagy and the CVT pathway require different levels of Atg1 kinase activity, or that Atg1 has different substrates under differing nutrient conditions. In higher eukaryotes, the role of Atg1 kinase activity may differ from that in yeast, as the CVT pathway has not been observed in metazoan cells. Consistent with the findings of Ohsumi and colleagues, however, the current results indicate that the kinase domain of Drosophila Atg1 is required for autophagy, since the kinase-inactive Atg1K38Q mutant failed to restore starvation-induced autophagy to Atg1 mutants, and was unable to induce autophagy when overexpressed. However, the partial reduction in size and TOR activity observed upon expression of Atg1K38Q, as well as its partial rescue of Atg1 mutants, indicate that Atg1 may have other, kinase-independent functions beyond regulating autophagy (Scott, 2007).
The mechanisms by which upstream signaling pathways regulate Atg1 are also likely to differ between yeast and multicellular organisms; essential components of the TOR-regulated Atg1 complex such as Atg13 and Atg17 are not readily identifiable in animal genomes. Nonetheless, the current results are consistent with a similar negative regulation of Atg1 by TOR, since activation of TOR signaling by Rheb overexpression or Tsc2 mutation suppresses the ability of Atg1 to induce autophagy. Increased Atg1 activity in response to loss of TOR signaling is likely to be critical for cell homeostasis and survival, since animals doubly mutant for Atg1 and Tor show a synthetic embryonic lethal phenotype. In addition, the results demonstrate an unexpected mode of signaling from Atg1 to TOR, in which increased Atg1 levels lead to downregulation of TOR kinase activity. It is unclear whether this reflects a direct effect of Atg1 on the activity of TOR or an upstream regulator, or an indirect consequence of the high level of autophagy in these cells, perhaps through increased turnover of TOR signaling components. The finding that a pool of TOR protein resides on intracellular vesicles and that TOR signaling is reduced in endocytic mutants suggests that TOR activity may respond to rates of vesicular trafficking, such as autophagy. Regardless of mechanism, these results suggest the existence of a self-reinforcing feedback loop, whereby increased Atg1 levels lead to downregulation of TOR activity, resulting in further activation of Atg1. In contrast, induction of autophagy in response to loss of TOR signaling is dampened by the resultant inactivation of S6K, which is required for normal autophagy. Thus TOR-mediated regulation of autophagy involves both positive and negative feedback (Scott, 2007).
Autophagy is a catabolic process that inversely correlates with cell growth, suggesting that increased levels of autophagy observed in growth-restricted cells may contribute to their reduced growth rate. The results presented here provide genetic support for this model. The data indicate that clones of autophagy-defective cells have a growth advantage over wild type cells under physiological conditions that normally induce autophagy, including starvation and rapamycin treatment. These results are confirmed by the marked size reduction of cells overexpressing Atg1, further supporting the role of autophagy as a negative effector of growth in TOR signaling. Thus, autophagy is partly responsible for the growth restriction resulting from physiological inhibition of TOR signaling, and is capable of inhibiting growth independent of TOR signaling (Scott, 2007).
In contrast to the growth advantage observed in autophagy defective cells, previous studies using yeast or cultured mammalian cells reported that inhibition of autophagy results in rapid cell death in response to starvation. This difference is likely due to the mosaic nature of the current experiments, in which clones of Atg1 mutant cells are surrounded by autophagy-competent wild type cells, and may in effect parasitize nutrients liberated through the autophagic activity of their neighbors. This is likely to be particularly evident in fat body cells, which are specialized for nutrient storage and mobilization. It is noted that this genetic mosaicism is similar to the situation facing tumor cells within wild type tissues. Thus, the increased growth capacity resulting from disruption of autophagy may contribute to the tumorigenicity of cells mutant for tumor suppressors such as PTEN, TSC1 & 2, beclin 1/Atg6 and possibly LC3/Atg8 and Atg7 (Scott, 2007 and references therein).
Whereas autophagy had an inhibitory effect on cell growth under physiological conditions, this was not the case in cells with severely disrupted TOR signaling, such as in cells with null mutations in Pdk1 Tor, despite the strong correlation of reduced cell growth and increased autophagy in these cells. Disruption of autophagy had no effect on the growth of Pdk1 null cells, and actually led to a further decrease in cell size of Tor mutants. In the complete absence of TOR signaling, nutrient uptake is severely curtailed, and under these extreme conditions the metabolic benefits of nutrients liberated by autophagy may outweigh its potential growth-inhibitory catabolic effect. It is concluded that autophagy has context-dependent effects on cell growth, providing for a rudimentary level of metabolism and growth under conditions of severe nutrient deprivation, acting as a net inhibitor of growth under conditions of reduced TOR signaling, and strongly inhibiting cell growth when induced to high levels. These findings add autophagy to the growing list of effector pathways and cellular processes through which TOR signaling controls cell growth, including translation, transcription, nutrient import and endocytosis (Scott, 2007).
Previous studies in a number of experimental systems indicate that the role of autophagy in cell death is also likely to be context-dependent. For example, autophagy has been found to protect against cell death in cases of growth factor withdrawal, starvation, and neurodegeneration, but to be required for some cases of autophagic cell death. Thus, observations of autophagic structures in dying cells are equally consistent with a causal, neutral or even inhibitory role of autophagy in cell death. The ability to induce autophagy through Atg1 overexpression has enabled a direct test of the potential role of autophagy in promoting cell death. The results indicate that induction of autophagy is sufficient to induce cell death. It was found that death resulting from Atg1-induced autophagy is suppressed by caspase inhibition and is associated with caspase activation, DNA fragmentation, and cytoskeletal disruption, suggesting that high levels of autophagy result in apoptotic cell death. The connection between apoptosis and autophagy is further supported by the recent demonstration that overexpression of the anti-apoptotic protein Bcl-2 can inhibit autophagy by interacting with the Atg6 homolog Beclin 1. Mutant versions of Beclin 1 that are unable to bind Bcl-2 stimulate autophagy and promote cell death, similar to the effects of Atg1 (Scott, 2007).
By what mechanisms might autophagy lead to cell death? The observation that starvation-induced autophagy is reversible and does not normally result in cell elimination suggests that starvation conditions may be protective against autophagy-induced death. Induction of autophagy is tolerated or even beneficial in cells with reduced biosynthetic activity, but may be detrimental in cells whose resources are devoted to continued growth. Self-limiting mechanisms also serve to prevent starvation-induced autophagy from proceeding at continuously high levels. During autophagic cell death, alternate activation of the autophagic machinery may circumvent these feedback mechanisms, resulting in high levels of sustained autophagy that are destructive to the cell. In addition, the level of Atg1 gene expression may be critical, since it is not upregulated under starvation conditions (Kirisako, 1999), but its levels peak at the beginning of the Drosophila pupal stage, when autophagic cell death is induced. Autophagic cell death may also result from the selective degradation of specific survival-promoting or death-inhibiting factors. In this regard, it was recently shown that the caspase-inhibitor zVAD results in targeted autophagic degradation of catalase, leading to accumulation of radical oxygen species and death (Scott, 2007).
In summary, these results demonstrate that constitutive induction of autophagy inhibits cell growth and leads to cell death, highlighting the importance of physiological mechanisms that restrain this process. In addition, the finding that Atg1 expression is sufficient to induce autophagy provides a new tool for experimental or therapeutic manipulation of autophagy. Compounds that target the kinase activity of Atg1 may lead to novel therapies for the wide range of diseases linked to autophagy (Scott, 2007).
Degradation of cytoplasmic components by autophagy requires the class III phosphatidylinositol 3 [PI(3)]-kinase Vps34, but the mechanisms by which this kinase and its lipid product PI(3) phosphate (PI(3)P) promote autophagy are unclear. In mammalian cells, Vps34, with the proautophagic tumor suppressors Beclin1/Atg6, Bif-1, and UVRAG, forms a multiprotein complex that initiates autophagosome formation. Distinct Vps34 complexes also regulate endocytic processes that are critical for late-stage autophagosome-lysosome fusion. In contrast, Vps34 may also transduce activating nutrient signals to mammalian target of rapamycin (TOR), a negative regulator of autophagy. To determine potential in vivo functions of Vps34, mutations were generated in the single Drosophila Vps34 orthologue (Phosphotidylinositol 3 kinase 59F), causing cell-autonomous disruption of autophagosome/autolysosome formation in larval fat body cells. Endocytosis is also disrupted in Vps34-/- animals, but this does not account for their autophagy defect. Unexpectedly, TOR signaling is unaffected in Vps34 mutants, indicating that Vps34 does not act upstream of TOR in this system. Instead, TOR/Atg1 signaling regulates the starvation-induced recruitment of PI(3)P to nascent autophagosomes. These results suggest that Vps34 is regulated by TOR-dependent nutrient signals directly at sites of autophagosome formation (Juhász, 2008).
Engulfment of cytoplasmic material into specialized double-membrane vesicles known as autophagosomes is the defining feature of a process referred to as macroautophagy or simply autophagy. Subsequent fusion of autophagosomes with the endolysosomal network leads to hydrolytic degradation of the sequestered material. This process provides eukaryotic cells with a mechanism for cytoplasmic renewal by which they can rid themselves of defective organelles and protein complexes. In addition, nonselective autophagy can be induced to high levels by starvation, providing an internal source of nutrients on which cells can survive extended periods of nutrient deprivation. Conversely, under some circumstances autophagy may be used as a killing mechanism, acting as an alternative or augmentation to apoptotic cell death. As autophagy has been implicated in several physiological and pathological conditions, including neurodegeneration, tumorigenesis, and aging, better understanding of the molecular mechanisms controlling autophagy and identification of pharmacological regulators of this process are important goals (Juhász, 2008).
Wortmannin and 3-methyladenine are well established inhibitors of autophagy. These compounds are broad-spectrum phosphatidylinositol 3 [PI(3)]-kinase inhibitors that disrupt autophagy by inhibiting Vps34, the enzymatic component of a multiprotein complex which also includes Vps15, Beclin1/Atg6, UVRAG, and Bif-1 in mammals and Vps15, Atg6, and Atg14 in yeast. Localized production of PI(3) phosphate (PI(3)P) by Vps34 can act to recruit proteins containing FYVE and PX domains to specific membrane compartment. In yeast, this Vps34 complex is critical for recruiting autophagy-related (Atg) proteins to the preautophagosomal structure, the yeast-specific site of autophagosome formation. The role of PI(3)P in autophagosome biogenesis is less well understood in higher eukaryotes, and whether it functions at the autophagosomal, the donor, or another membrane has not been determined (Juhász, 2008).
Vps34 is also required more broadly for several vesicular trafficking processes that may have indirect impacts on autophagy. These include sorting of hydrolytic enzymes to the lysosome/vacuole and early steps in the endocytic pathway. In mammalian cells, autophagosomes have been shown to fuse with early or late endosomes before fusion with lysosomes, resulting in intermediate structures known as amphisomes. Recently, mutations in components of the endosomal sorting complex required for transport (ESCRT) complex, which is required for the transition from early to late (multivesicular) endosomes, have been shown to block autophagy by inhibiting autophagosome-endosome fusion. Thus, the effect of PI(3)-kinase inhibitors on autophagy may be due, in part, to these more general trafficking functions of Vps34 (Juhász, 2008).
Recent work has shown that Vps34 can also function in a nutrient-sensing pathway upstream of the target of rapamycin (TOR) in several mammalian cell lines. Disruption of Vps34 activity with blocking antibodies or siRNA was found to inhibit activation of TOR by insulin, amino acids, and glucose. As TOR signaling inhibits autophagy, these findings are at odds with the conserved role of Vps34 in promoting autophagy under starvation conditions, suggesting that distinct complexes or pools of Vps34 may be subject to different modes of regulation (Juhász, 2008).
This paper addresses how these multiple potential roles of Vps34 are coordinated to regulate autophagy in Drosophila. The findings suggest that despite a critical role for Vps34 in endocytic uptake and recycling, its primary function in autophagy in vivo is limited to its direct role at the nascent autophagosome. Vps34 is not required for TOR activity in this system, and starvation results in a TOR/Atg1-dependent recruitment of Vps34 activity to the autophagosomal membrane (Juhász, 2008).
Previous work in mammalian and yeast systems has identified a wide range of vesicle trafficking processes regulated by Vps34, including autophagy, endocytosis, endosome maturation, and both anterograde and retrograde trafficking between the Golgi and lysosome. The current findings indicate that despite these activities, the in vivo role of Vps34 in autophagy is largely limited to its function at the autophagosome. Although fluid-phase endocytosis, endocytic recycling of Notch, and trafficking of lysosomal proteins are disrupted by mutation of Vps34, the results suggest that events subsequent to autophagosome formation, including fusion between autophagosomes and endosomes or lysosomes and subsequent lysosomal degradation, are not rate limiting in the absence of Vps34. Why does endocytic disruption lead to autophagosome fusion defects in ESCRT mutants but not in Vps34 mutants? Accumulation of endocytic tracer at the periphery of Vps34 mutant cells suggests that Vps34 functions at an early step of endocytosis, and apparently this event, as well as normal endocytic flux is not essential for fusion of autophagosomes with elements of the endosomal-lysosomal compartment. Interestingly, the accumulation of autophagosomes in ESCRT/Vps34 double mutants indicates that loss of Vps34 does not completely prevent autophagosome formation. Similarly, the lack of autophagosome accumulation in Vps34 single mutants indicates that ESCRT complexes are at least partially functional in the absence of Vps34. Thus, PI(3)P may not be absolutely essential for these processes, or perhaps sufficient levels of PI(3)P are generated independently of Vps34 by the class II PI(3) kinase or by PI(3,4)P or PI(3,5)P phosphatases. Since ESCRT components are required for multivesicular body formation but not autophagy in yeast, it will be interesting to determine whether the requirement for ESCRT complexes in autophagy in higher eukaryotes reflects their role in multivesicular body formation or an alternate function (Juhász, 2008).
The cellular compartment in which Vps34 acts to promote autophagy and the mechanisms by which it is regulated by nutrient signals have remained unresolved. In mammalian cells, Beclin1/Atg6 has been reported to localize to the trans-Golgi network, ER, and mitochondria. It was recently shown that Beclin1-Vps34 complexes can be inhibited by the antiapoptotic factor Bcl-2 in a nutrient-dependent manner. Bcl-2 mutants that are targeted to the ER, but not to mitochondria, retain their capacity to inhibit starvation-induced autophagy, suggesting that the ER is an important site of Beclin1-Vps34 regulation. However, it is unknown how these organelles contribute to the formation of autophagosomes, and recent studies suggest that rather than budding off a preexisting compartment, the autophagosomal membrane is likely to form de novo from small lipid transport vesicles or lipoprotein complexes. The finding that myc-2xFYVE is recruited to GFP-Atg8a-positive structures under starvation conditions indicates that Vps34 activity is targeted directly to autophagosomes in a TOR/Atg1-dependent manner. Although these results do not distinguish between a role for TOR/Atg1 signaling in regulating Vps34 activity versus providing a platform on which Vps34 complexes can assemble, together with these previous studies they indicate that Vps34 is likely to promote autophagy by different mechanisms from multiple cellular locations (Juhász, 2008).
How the TOR signaling pathway senses intracellular levels of nutrients, such as amino acids, has been poorly understood despite considerable work in yeast, mammalian, and other model systems. The recent identification of Vps34 as a transducer of this signal in mammalian cells thus represents a significant new insight into this issue. However, further work is necessary to determine the extent to which this mechanism is generally conserved, since starvation appears to have opposing effects on Vps34 activity in different cell types. The results presented in this study fail to support this model in Drosophila; mutation of Vps34 does not appear to influence TOR-dependent phenotypes nor to disrupt TOR-dependent signaling. This may reflect a fundamental difference in signaling mechanisms between the fly and mammalian systems. The makeup of Vps34 complexes has diverged significantly between yeast and metazoans, and perhaps components of this complex, such as Ambra1, that appear to be unique to vertebrates may confer functions not found in flies. It is also possible that production of PI(3)P by Vps34-independent mechanisms is more efficient in D. melanogaster than in mammalian cells and, thus, a role for Vps34 in TOR signaling may be obscured by these other sources. The continued ESCRT function and basal level of autophagy in Vps34 null mutants are consistent with this possibility. Alternatively, the current findings may reflect important differences between the roles of TOR and Vps34 in vivo versus in cultured cells as well as the experimental paradigms of these systems. For example, although complete starvation is commonly used to inactivate TOR in cell culture studies, such experiments may not accurately mimic physiologically relevant events, given the inherent capacity of intact organisms to buffer changes in nutrient levels. Additional studies in an in vivo mammalian system will be helpful to clarify these issues (Juhász, 2008).
The TOR kinases are conserved negative regulators of autophagy in response to nutrient conditions, but the signaling mechanisms are poorly understood. This study describes a complex containing the protein kinase Atg1 and the phosphoprotein Atg13 that functions as a critical component of this regulation in Drosophila. Knockout of Atg1 or Atg13 results in a similar, selective defect in autophagy in response to TOR inactivation. Atg1 physically interacts with TOR and Atg13 in vivo, and both Atg1 and Atg13 are phosphorylated in a nutrient-, TOR- and Atg1 kinase-dependent manner. In contrast to yeast, phosphorylation of Atg13 is greatest under autophagic conditions and does not preclude Atg1-Atg13 association. Atg13 stimulates both the autophagic activity of Atg1 and its inhibition of cell growth and TOR signaling, in part by disrupting the normal trafficking of TOR. In contrast to the effects of normal Atg13 levels, increased expression of Atg13 inhibits autophagosome expansion and recruitment of Atg8/LC3, potentially by decreasing the stability of Atg1 and facilitating its inhibitory phosphorylation by TOR. Atg1/Atg13 complexes thus function at multiple levels to mediate and adjust nutrient-dependent autophagic signaling (Chang, 2009).
The remarkable conservation of autophagy-related proteins and processes between yeast and higher eukaryotes has contributed to a rapid advance in understanding of this process in multicellular animals. The data presented in this study extend this conservation to the mechanisms of autophagy regulation by nutrient-dependent TOR signaling. As in yeast, Drosophila Atg13 forms a complex with Atg1, stimulates its activity and is highly phosphorylated in a nutrient and TOR-dependent manner. Although the relevant substrates of Atg1 remain to be identified, these results suggest that this fundamental pathway from nutrient signal to autophagic induction has
been largely retained between yeast and metazoans. This regulatory link between TOR and
Atg1/Atg13 thus represents one of the most highly conserved TOR outputs described to date.
Despite this conservation, critical differences were identified in the behaviors of these
proteins, which likely stem in part from differences in the physiology of lower vs. higher
eukaryotes. An essential role
in autophagy has been reported for human Atg13 using RNAi-mediated knockdown in cultured cell lines, and interactions were described between Atg13 and Ulk1 and Ulk2 similar to those reported in this study. Others have also described related interactions between mammalian Atg13 and Ulk kinases. Together, these studies
point to a model whereby Atg13 is phosphorylated and interacts with Atg1 under both fed and starved conditions in metazoans, in contrast to the growth-dependent phosphorylation and dissociation of Atg13/Atg1 observed in yeast. Although the greater nutrient buffering capacity of multi- vs. unicellular animals probably contributes to these differences, the requirement for significant basal rates of autophagy in the maintenance of large, long-lived metazoan cells may dictate that the Atg1-Atg13 complex remain partially active even under fed conditions. Furthermore, although Atg1 can clearly associate with phosphorylated Atg13, dephosphorylation of TOR-dependent sites on Atg13 may be masked by increased phosphorylation by Atg1, but may nonetheless contribute to regulation of Atg1-Atg13 interaction or activity. The observation that Atg1 kinase activity plays a role under both fed and starved conditions in supporting hyperphosphorylation of Atg13 suggests that rather than switching Atg1 kinase between off and on states, nutrient signals may instead affect its substrate specificity or accessibility (Chang, 2009).
Metazoan Atg1/Atg13 complexes have also accrued additional regulatory mechanisms
that have not been described in yeast, including negative feedback from Atg1 to TOR,
phosphorylation of Atg13 by Atg1, and nutrient-dependent effects on the stability of these
proteins. These additional layers of regulation are in keeping with the elaboration of intracellular signaling pathways in metazoans. The observation that Atg13 phosphorylation is both TOR- and Atg1-dependent under fed conditions suggests a model whereby phosphorylation by one of these kinases may serve as a priming event for the other. In addition, the role of Atg1-dependent phosphorylation of Atg13 under starvation conditions remains an important question. Whereas the Atg1-independent localization of Atg13 to autophagosomes and its activation of Atg1 indicate that Atg13 functions upstream of Atg1, the finding that Atg13 acts as a substrate for Atg1-dependent phosphorylation raises the possibility that Atg13 may also act to transduce signals downstream of Atg1. Identification of additional Atg1 substrates and Atg13 interacting
proteins may help clarify this issue (Chang, 2009).
Given its positive role in autophagy induction, the ability of Atg13 to inhibit autophagy when overexpressed was unexpected. This may in part reflect a dominant negative effect of Atg13 overexpression causing titration of Atg1 complexes or competition with other Atg1 substrates. However, the observation that Atg13 stimulates TOR-dependent phosphorylation of Atg1 and that Atg1 levels increase in Atg13 mutant cells suggests that Atg13 has both positive and negative roles in autophagy induction. These opposing activities of Atg13 are reminiscent of the mTOR complex I component Raptor, which plays an essential role in TOR signaling yet also inhibits TOR activity under starvation conditions. The results suggest that
Atg13 may play an analogous role, switching between states of promoting or inhibiting
autophagy, thereby sharpening the response to changes in nutrient conditions. These findings suggest that the relative ratio of Atg13 to Atg1 or to other components of the complex may play an important role in dictating its activity. In this regard, the differential regulation of Atg1 and Atg13 levels by TOR could provide an additional mechanism whereby TOR signaling can influence autophagic activity. Whereas the autophagy-defective phenotypes of Atg1 and Atg13 mutants reveals the dominant positive roles of these genes, disruption of other putative components of this complex leads to autophagy induction, suggesting some components may play primarily negative roles (Chang, 2009).
In conclusion, the results demonstrate that Atg1/Atg13 complexes play an essential,
conserved role in promoting autophagy in response to TOR inactivation, with additional
regulatory functions unique to metazoans. Further insight into the regulation of this complex and identification of its targets may lead to additional means of manipulating autophagy rates for therapeutic purposes (Chang, 2009).
The TOR pathway mediates nutrient-responsive regulation of cell growth and metabolism in animals. TOR Complex 1 activity depends, among other things, on amino acid availability. MAP4K3 was recently implicated in amino-acid signaling in cell culture. This study reports the physiological characterization of MAP4K3 mutant flies. Flies lacking MAP4K3 have reduced TORC1 activity detected by phosphorylation of S6K and 4EBP. Furthermore MAP4K3 mutants display phenotypes characteristic of low TORC1 activity and low nutrient availability, such as reduced growth rate, small body size, and low lipid reserves. The differences between control and MAP4K3 mutant animals diminish when animals are reared in low-nutrient conditions, suggesting that the ability of TOR to sense amino acids is most important when nutrients are abundant. Lastly, physical interaction is shown between MAP4K3 and the Rag GTPases raising the possibility they might be acting in one signaling pathway (Bryk, 2010).
The multiprotein complex TORC1, containing TOR kinase, is a central regulator of cellular growth and metabolism in animals. It is activated by a number of inputs relating to cellular energy and nutrient status. These include insulin, glucose, cellular energy levels and amino acid availability. In response, TORC1 activates protein synthesis via a number of mechanisms including activation of the ribosomal S6 kinase (S6K), repression of the translational inhibitor 4E-BP, and promotion of ribosome biogenesis via myc. In particular since TORC1 is a master regulator of protein biosynthesis, its regulation by amino acids, the building blocks of proteins, likely constitutes an important regulatory feedback mechanism. Furthermore, the importance of amino acid signaling to TOR is highlighted by the observation that circulating amino acids are elevated in humans with obesity, where they have been shown to activate TORC1 activity and modulate glucose metabolism. Despite this, understanding of the molecular mechanism by which amino acids regulate TOR remains fragmentary (Bryk, 2010).
Three protein complexes have recently been implicated in the activation of TORC1 in response to amino acids. The human class III PI3K (phosphoinositide 3-kinase) hVps34 is activated by amino acids via a calcium dependent mechanism (Gulati, 2008). This leads to accumulation of phosphatidylinositol 3-phosphate (PI(3)P) in cells, which is thought to cause the recruitment of proteins recognizing PI(3)P to early endosomes, forming an intracellular signaling platform that leads to TORC1 activation. This feature of the pathway may be specific for vertebrates, as flies mutant for Vps34 have been reported to not have TORC1 signaling defects. Recently, two groups discovered that Rag GTPases mediate amino acid signaling to TORC1. The emerging picture is that amino acids change the GDP/GTP loading of the Rag GTPases, thereby stimulating the binding of Rag heterodimeric complexes to TORC1. This in turn causes TORC1 to change its intracellular localization, perhaps relocalizing it to vesicles containing the activator Rheb. This mechanism appears to be evolutionarily conserved from flies to humans (Bryk, 2010).
The third protein recently identified as a mediator of amino acid signaling to TOR is MAP4K3 (Findlay, 2007); in HeLa cells the kinase activity of MAP4K3 is activated in the presence of amino acids. In turn, MAP4K3 is required for TOR to phosphorylate its targets S6K and 4E-BP1 in response to amino acid sufficiency. The cell-culture data also suggest this mechanism is conserved from flies to humans as knockdown of Drosophila MAP4K3 causes a reduction in TOR activity (Bryk, 2010).
Although considerable progress has been made, important questions remain unanswered. The role of TORC1 activity in vivo has been well studied in flies and mice, but fundamental issues regarding the regulation of TOR by amino acids have thus far only been explored in vitro in cell culture. To assess the functional significance of the ability of TORC1 to sense amino acids in the organismal context, use has been made of a Drosophila mutant for MAP4K3 (CG7097). dMAP4K3
mutant flies have reduced TOR activity, detected by phosphorylation of TOR targets. dMAP4K3 mutants are viable but display physiological aberrations emblematic of animals starved of nutrients: MAP4K3 mutant flies have retarded growth, reduced size, and low lipid reserves. Both the biochemical results and the phenotypes indicate that MAP4K3 modulates, but is not absolutely required, for TOR activity in vivo. This is similar to what is observed with other modulators of the pathway, such as Melted. Unexpectedly, the function of MAP4K3 is most required when nutrient conditions are rich (Bryk, 2010).
Recent reports have shown that not all components identified in cell culture as regulators of TORC1 activity also affect TORC1 in vivo in an animal model. The purpose of this study was two-fold: (1) to analyze whether MAP4K3 regulates TORC1 activity in vivo in the fly, and (2) to study the physiological consequences for the organism when the ability of TORC1 to sense amino acids is impaired (Bryk, 2010).
Biochemical evidence is presented that TORC1 activity is reduced in MAP4K3 mutant animals, consistent with published cell-culture data showing that MAP4K3 is required for full TORC1 activation (Findlay, 2007). Furthermore, MAP4K3 mutants have defects typical of reduced TORC1 activity. They are delayed in their development due to a reduced rate of growth. They eventually pupate leading to adults of reduced size and their tissues are comprised of cells that are smaller than normal. Furthermore, MAP4K3 mutants have significantly reduced triglyceride stores compared to controls. These physiological effects are similar to the phenotypes observed with mutants for other regulators of TOR, such as Melted. Melted mutant flies are also 10% smaller than controls and are significantly leaner (Bryk, 2010).
As a whole, the MAP4K3- mutant phenotypes emulate the physiological effects observed when flies are grown on conditions of limiting food. When wildtype larvae are put on a low-nutrient diet, they are delayed in pupation and yield animals of small size that are lean. Thus loss of MAP4K3 activity phenocopies a reduced nutrient environment, consistent with MAP4K3 playing a role in the ability of animals to sense their nutrient conditions. This suggests the ability of TORC1 to sense amino acids is most important when nutrient conditions are rich, allowing animals to accelerate their growth accordingly. In contrast, on a low-nutrient diet, control and MAP4K3 mutant flies grow equally slowly consistent with TOR activity being low in both groups. This parallels nicely the results reported in cell culture by (Findlay, 2007): In the absence of amino acids, both control and MAP4K3 knockdown cells have low TOR activity whereas in the presence of amino acids, TOR is activated strongly in control cells but only weakly in MAP4K3 knockdown cells (Bryk, 2010).
Unexpectedly, it was found that MAP4K3 mutant animals are viable, although they have an elevated mortality rate compared to controls. This suggests that the amino acid sensing pathway might only modulate TORC1 activity. If TORC1 activity were completely blunted in MAP4K3 mutants, the animals would be dead, as is the case for TOR or Rheb mutants. Consistent with this, residual TORC1 activity was observed in MAP4K3 mutants, as detected by phosphorylation of the TORC1 targets S6K and 4EBP. This also parallels results from cell culture. The results presented in Findlay (2007) are obtained with cells starved of serum and consequently of insulin signaling. In the presence of insulin signaling, which resembles the physiological situation more closely, MAP4K3 mutant cells still retain residual TOR activity, similar to what was observed in vivo in this study. Consistent with these findings, it was observed that the Rheb expression is able to drive tissue growth also in the absence of MAP4K3 (Bryk, 2010).
Both MAP4K3 and the Rag GTPases have recently been shown to be required for amino acids to stimulate TORC1 activity. While studying dMAP4K3, it was noticed that dMAP4K3 binds physically to the Rag GTPases, suggesting they might act together as components of a single signaling pathway. This interaction is likely specific for several reasons: (1) no binding of MAP4K3 to another GTPase, Rheb, could be detected, (2) binding of MAP4K3 to the RagA/C complex was significantly stronger than binding of an unrelated HA-tagged protein, HA-medea (3) MAP4K3 bound FLAG-RagC significantly stronger than FLAG-RagA showing that MAP4K3 distinguishes between two Rag proteins and (4) binding of MAP4K3 to RagC depended on its GDP/GTP state (Bryk, 2010).
Further studies will be required to test whether this interaction is important for TORC1 to sense amino acids. The data suggest that MAP4K3 might be functioning upstream of the Rag GTPases, and not downstream since activated RagA does not require MAP4K3 to promote tissue growth in vivo. This raises the possibility that the Rag GTPases may be substrates for MAP4K3 phosphorylation. Indeed, RagC is phosphorylated in vivo in Kc167 cells on Ser388. If MAP4K3 were to phosphorylate RagC, this would provide a mechanism for regulation of the Rag GTPases, which to date is mysterious. Work in the near future should shed further light on this issue (Bryk, 2010).
In summary, this study has characterized the physiological function of MAP4K3 in Drosophila, and shown that it modulates TORC1 activity, tissue growth and lipid metabolism in the animal. Physical interaction data hints at a possible link between MAP4K3 and the Rag GTPases. The organismal function of amino acid sensing by TORC1 is mainly required to spur growth when nutrient conditions are rich (Bryk, 2010).
MAP4K3 is a conserved Ser/Thr kinase that has being found in connection with several signalling pathways, including the Imd, EGFR, TORC1 and JNK modules, in different organisms and experimental assays. This study analyzed the consequences of changing the levels of MAP4K3 expression in the development of the Drosophila wing, a convenient model system to characterize gene function during epithelial development. Using loss-of-function mutants and over-expression conditions it was found that MAP4K3 activity affects cell growth and viability in the Drosophila wing. These requirements are related to the modulation of the TORC1 and JNK signalling pathways, and are best detected when the larvae grow in a medium with low protein concentration (TORC1) or are exposed to irradiation (JNK). MAP4K3 was also shown to display strong genetic interactions with different components of the InR/Tor signalling pathway, and can interact directly with the GTPases RagA and RagC and with the multi-domain kinase Tor. It is suggested that MAP4K3 has two independent functions during wing development, one related to the activation of the JNK pathway in response to stress and other in the assembling or activation of the TORC1 complex, being critical to modulate cellular responses to changes in nutrient availability (Resnik-Docampo, 2011).
This study has characterised the consequences of changing the amount of MAP4K3, encoded by hppy, in the development of the wing disc, focussing on its relationships with the TORC1 signalling pathway. Previous data suggested that MAP4K3 might be related with a variety of signalling pathways, including EGFR, ImD, JNK and TOR. For these reasons, the advantages of the wing model was used to analyse hppy, as in this system changes in the level of signalling by a variety of pathways lead to pathway-specific phenotypes. A reduction of hppy expression in the wing, using interference RNA or loss-of-function alleles, did not uncover a critical requirement of the gene for embryonic or larval viability. In hppy mutant wings, only a weak reduction was found in wing size and cell size, compatible with a moderate reduction of TORC1 activity. It has been recently reported (Bryk, 2010) that the developmental delay caused by protein starvation is similar in wild type and hppy mutant larvae, suggesting that MAP4K3 is required in vivo to activate TOR and promote growth mostly when amino acid conditions are rich. In contract, this study found a significant requirement for the gene when hppy mutant larvae grow under starvation conditions. Thus, these flies still develop smaller wings than controls, indicating a functional requirement of hppy when the availability of proteins is reduced. This difference could be due to the parameters measured (developmental delay vs. cell and wing size) or to the remnants of hppy function in the alleles used in each experiment (Resnik-Docampo, 2011).
It was also found that, loss of hppy does not affect cell viability or JNK signalling, but that in a hppy loss-of-function genetic background the activation of JNK signalling in response to irradiation is reduced. Thus, the function of hppy might become significant mostly when the organism is challenged by stress signals induced for example by irradiation, indicating a role for the gene in the modulation of JNK signalling in vivo (Resnik-Docampo, 2011).
The increase in happyhour expression does have more dramatic consequences than its loss, causing a severe reduction in the size of the wing independently of environmental conditions. Wing size reduction is associated with both apoptosis and a smaller than normal cell size. The overall morphology and pattern of these wings is normal, with only a weak phenotype of extra-veins in the strongest combinations. Cell death induction and reduced cell size are the diagnostic phenotypes of increased JNK and reduced InR/Tor signalling, respectively. The same processes are affected by loss of MAP4K3 expression in the wing, and therefore, from this analysis it is concluded that MAP4K3 has the potential to activate cell death through the JNK signalling pathway, and also that it can interfere with some component/s of the InR/Tor cascade. The effects of loss- and gain of MAP4K3 on JNK activity are opposite, which is expected from a protein with kinase activity. In contrast both loss and gain of MAP4K3 seem to reduce the function of TORC1. It is likely that in this case MAP4K3 acts as part of a protein complex that can be made non-functional by changes in the stochiometry of its components. What seems clear is that the effects of MAP4K3 on JNK and TORC1 are exerted through independent mechanisms, because the contribution of cell death to the wing phenotype of MAP4K3 over-expression is very modest, and Tor reductions only lead to cell death when cells with different levels of Tor activity are confronted (Resnik-Docampo, 2011).
The phenotype of MAP4K3 over-expression is very sensitive to changes in the levels or activity of several members of the InR/Tor pathway. Thus, strong synergistic interactions were observed when Akt, raptor and Tor are reduced in the background of MAP4K3 over-expression, and the presence of the dominant-negative form TorTED in this background entirely eliminates the wing. Conversely, loss of hppy expression rescues the effects of TorTED expression. These results suggest that MAP4K3 could act at the level of TORC1. This possibility is compatible with the suppression by MAP4K3 over-expression of phenotypes caused by increased levels of InR/Tor signalling generated by lower than normal levels of PTEN and TSC1/2. In addition to genetic data in the wing, experiments in cell culture with both the fly and human MAP4K3 homologue proteins indicated that MAP4K3 is required to generate maximal activity of TORC1 in response to aminoacids. Therefore, it is suggested that although MAP4K3 is normally required to promote TORC1 signalling, when the protein is over-expressed the balance between TORC1 components required for its normal function in vivo is modified. This effect appears to depend exclusively on the kinase domain of MAP4K3, because the over-expression of this domain causes a strong reduction in wing and cell size. This sstudy has shown that MAP4K3 can interact with RagA, RagC and Tor in pull-down experiments in vitro, and therefore it is speculated that the excess of MAP4K3 alters the phosphorylation levels of TORC1 components and this leads to the assembly of inactive complexes. A similar mechanism might explain the dominant-negative effect of Tor, as it was suggested that Tor over-expression leads to the sequestering of TORC1 components in non-functional complexes. In summary, it is suggested that MAP4K3 normally potentiate TORC1 and JNK functions in response to environmental challenges, without being strictly required to generate some levels of TORC1 or JNK activity, and that MAP4K3 hyper-activity leads to high levels of JNK signalling and to reduced TORC1 function, in this case due to the formation of inactive TORC1 complexes (Resnik-Docampo, 2011).
The circadian clock coordinates cellular and organismal energy metabolism. The importance of this circadian timing system is underscored by findings that defects in the clock cause deregulation of metabolic physiology and result in metabolic disorders. On the other hand, metabolism also influences the circadian clock, such that circadian gene expression in peripheral tissues is affected in mammalian models of obesity and diabetes. However, to date there is little to no information on the effect of metabolic genes on the central brain pacemaker which drives behavioral rhythms. This study found that the AKT and TOR-S6K pathways, which are major regulators of nutrient metabolism, cell growth, and senescence, impact the brain circadian clock that drives behavioral rhythms in Drosophila. Elevated AKT or TOR activity lengthens circadian period, whereas reduced AKT signaling shortens it. Effects of TOR-S6K appear to be mediated by SGG/GSK3beta, a known kinase involved in clock regulation. Like SGG, TOR signaling affects the timing of nuclear accumulation of the circadian clock protein Timeless. Given that activities of AKT and TOR pathways are affected by nutrient/energy levels and endocrine signaling, these data suggest that metabolic disorders caused by nutrient and energy imbalance are associated with altered rest:activity behavior (Zheng, 2010).
There are several possible mechanisms by which nutrient and energy metabolism could affect peripheral clocks. Local physiological factors dependent on metabolic activity could influence the expression of core clock components and of nuclear receptors that regulate clock gene expression. Indeed, cellular redox state, AMPK activity, NAD+ levels, and SIRT1 activities appear to feed into the circadian clock in peripheral tissues such as the liver. AMPK, which acts upstream of TSC in mammals, directly phosphorylates Cryptochrome in peripheral tissues. However, prior to this work, there was no known mechanism for the modulation of the central pacemaker by nutrient-sensing pathways. This study identifies such a mechanism by demonstrating that metabolic genes such as AKT and TOR-S6K act in the central pacemaker cells in the brain. The lengthened circadian period caused by high-fat diet in mammals is likely mediated by these molecules. This conclusion is further supported by a recent cell-culture-based genome-wide RNAi study that implicated the PI3K-TOR pathway in the regulation of circadian period. In addition, another ribosomal S6 kinase (S6KII) was found to influence the circadian clock through its interaction with casein kinase 2β. Importantly, daily fasting:feeding cycles driven by the central clock regulate circadian gene transcription in the liver, whereas clock function in the liver contributes to energy homeostasis. It is speculated that metabolic stress or energy imbalance affects AKT and TOR-S6K signaling, resulting in general circadian disruption, which in turn exacerbates metabolic deregulation and, consequently, facilitates the development of metabolic syndromes prevalent in modern society (Zheng, 2010).
Genetic studies in Drosophila reveal an important role for Myc in controlling growth. Similar studies have also shown how components of the insulin and target of rapamycin (TOR) pathways are key regulators of growth. Despite a few suggestions that Myc transcriptional activity lies downstream of these pathways, a molecular mechanism linking these signaling pathways to Myc has not been clearly described. Using biochemical and genetic approaches this study tried to identify novel mechanisms that control Myc activity upon activation of insulin and TOR signaling pathways.
Biochemical studies show that insulin induces Myc protein accumulation in Drosophila S2 cells, which correlates with a decrease in the activity of glycogen synthase kinase 3-β (GSK3β) a kinase that is responsible for Myc protein degradation. Induction of Myc by insulin is inhibited by the presence of the TOR inhibitor rapamycin, suggesting that insulin-induced Myc protein accumulation depends on the activation of TOR complex 1. Treatment with amino acids that directly activate the TOR pathway results in Myc protein accumulation, which also depends on the ability of S6K kinase to inhibit GSK3β activity. Myc upregulation by insulin and TOR pathways is a mechanism conserved in cells from the wing imaginal disc, where expression of Dp110 and Rheb also induces Myc protein accumulation, while inhibition of insulin and TOR pathways result in the opposite effect. Functional analysis, aimed at quantifying the relative contribution of Myc to ommatidial growth downstream of insulin and TOR pathways, revealed that Myc activity is necessary to sustain the proliferation of cells from the ommatidia upon Dp110 expression, while its contribution downstream of TOR is significant to control the size of the ommatidia. This study presents novel evidence that Myc activity acts downstream of insulin and TOR pathways to control growth in Drosophila. At the biochemical level it was found that both these pathways converge at GSK3β to control Myc protein stability, while genetic analysis shows that insulin and TOR pathways have different requirements for Myc activity during development of the eye, suggesting that Myc might be differentially induced by these pathways during growth or proliferation of cells that make up the ommatidia (Parisi, 2011).
Previous studies in vertebrates have indicated a critical function for Myc downstream of growth factor signaling including insulin-like growth factor, insulin and TOR pathways. In Drosophila, despite a few notes that Myc transcriptional activity acts downstream of insulin and TOR pathways, no clear molecular mechanisms linking these pathways to Myc have been elucidated yet (Parisi, 2011).
It has been demonstrated that inhibition of GSK3β prevents Myc degradation by the proteasome pathway. This study further unravels the pathways that control Myc protein stability and shows that signaling by insulin and TOR induce Myc protein accumulation by regulating GSK3β activity in S2 cells. GSK3β is a constitutively active kinase that is regulated by multiple signals and controls numerous cellular processes. With the biochemical data it is proposed that GSK3β acts as a common point where insulin and TOR signaling converge to regulate Myc protein stability (see Model showing the proposed relationship between Myc and the insulin and TOR signaling pathways). In particular, activation of insulin signaling was shown to induce activation of Akt, an event that is accompanied by GSK3β phosphorylation on Ser 9 that causes its inactivation and Myc protein to stabilize. Interestingly, insulin-induced Myc protein accumulation, when GSK3β activity was blocked by the presence of LiCl or by expression of GSK3β-KD, was similar to that obtained with insulin alone. Since it was shown that activation of insulin signaling leads to GSK3β inhibition and to an increase in Myc protein, if insulin and GSK3β signaling were acting independently, it would be expected that activation of insulin signaling concomitantly with the inhibition of GSK3β activity would result in a higher level of Myc than that obtained with insulin or LiCl alone. The results instead showed a similar level of Myc protein accumulation with insulin in the presence of GSK3β inhibitors as compared to insulin alone, supporting the hypothesis that GSK3β and insulin signaling, at least in the current experimental condition, depend on each other in the mechanism that regulates Myc protein stability (Parisi, 2011).
In a similar biochemical approach, the effect of AAs was analyzed on Myc protein stability and how TOR signaling is linked to mechanisms that inactivate GSK3β to stabilize Myc protein in S2 cells. In these experiments it was possible to demonstrate that treatment with amino acids (AAs) increased Myc protein stability, and it was also shown that treatment with rapamycin, an inhibitor of TORC1, reduced insulin-induced Myc upregulation. The reduction of Myc protein accumulation by rapamycin was blocked by inhibition of the proteasome pathway, linking TOR signaling to the pathway that controls Myc protein stability. TORC1 is a central node for the regulation of anabolic and catabolic processes and contains the central enzyme Rheb-GTPase, which responds to amino acids by activating TOR kinase to induce phosphorylation of p70-S6K and 4E-BP1. Analysis of the molecular mechanisms that act downstream of TOR to regulate Myc stability shows that AA treatment induces p70-S6K to phosphorylate GSK3β on Ser 9, an event that results in its inactivation and accumulation of Myc protein (Parisi, 2011).
Reducing GSK3β activity with LiCl, in medium lacking AAs, resulted in a slight increase in Myc protein levels. Adding back AAs lead to a substantial increase in Myc protein levels, which did not further increase when AAs where added to cells in the presence of the GSK3β inhibitor LiCl. These events were accompanied by phosphorylation of S6K on Thr 398, which correlated with phosphorylation of GSK3β on Ser 9. From these experiments it is concluded that TOR signaling also converges to inhibit GSK3β activity to regulate Myc protein stability. However, it has to be pointed out that since AAs alone increased Myc protein levels to a higher extent than that observed with LiCl alone, the experiments also suggest that Myc protein stability by TOR signaling is not solely directed through the inhibition of GSK3β activity, but other events and/or pathways contribute to Myc regulation. In conclusion, the biochemical experiments demonstrate that GSK3β acts downstream of insulin and TOR pathways to control Myc stability, however it cannot be excluded that other pathways may control Myc protein stability upon insulin and amino acids stimulation in S2 cells (Parisi, 2011).
Reduction of insulin and TOR signaling in vivo reduces cell size and proliferation, and clones mutant for chico, the Drosophila orthologue of IRS1-4, or for components of TOR signaling, are smaller due a reduction in size and the number of cells. The experiments showed that reducing insulin signaling by expression of PTEN or using TORTED, a dominant negative form of TOR, decreased Myc protein levels in clones of epithelial cells of the wing imaginal discs, while the opposite was true when these signals were activated using Dp110 or RhebAV4 . Those experiments suggested that the mechanism of regulation of Myc protein by insulin and TOR pathways was conserved also in vivo in epithelial cells of the larval imaginal discs (Parisi, 2011).
During these experiments it was also noted that Myc protein was induced in the cells surrounding and bordering the clones (non-autonomously), particularly when clones where positioned along the dorsal-ventral axis of the wing disc. This upregulation of Myc protein was not restricted to components of the insulin signaling pathway since it was also observed in cells surrounding the clones mutant for components of the Hippo pathway or for the tumor suppressor lethal giant larvae (lgl), which upregulates Myc protein cell-autonomously. It is suspected that this non-autonomous regulation of Myc may be induced by a novel mechanism that controls proliferation of cells when 'growth' is unbalanced. It can be speculated that clones with different growth rates, caused by different Myc levels, might secrete factors to induce Myc expression in neighboring cells. As a consequence, these Myc-expressing cells will speed up their growth rate in an attempt to maintain proliferation and tissue homeostasis. Further analysis is required to identify the mechanisms responsible for this effect (Parisi, 2011).
In order to distinguish if Myc activity was required downstream of insulin and TOR signaling to induce growth, a genetic analysis was performed. The ability to induce growth and proliferation was measured in the eye by measuring the size and number of the ommatidia from animals expressing members of the insulin and TOR pathways in different dm genetic background (dm+, dmP0 and dm4). The data showed that Dp110 increased the size and number of the ommatidia, however only the alteration in the total number was dependent on dm levels. These data suggest that Myc is required downstream of insulin pathway to achieve the proper number of ommatidia. However, when insulin signaling was reduced by PTEN, a significant decrease in the size of ommatidia was seen and it was dependent on dm expression levels, suggesting that Myc activity is limiting for ommatidial size and number. Activation of TOR signaling induces growth, and the genetic analysis showed that Myc significantly contributes to the size of the ommatidial cells thus suggesting that Myc acts downstream of TOR pathway to control growth (Parisi, 2011).
Recent genomic analysis showed a strong correlation between the targets of Myc and those of the TOR pathway, implying that they may share common targets. In support of this observation, mosaic analysis with a repressible cell marker (MARCM) experiments in the developing wing disc showed that overexpression of Myc partially rescues the growth disadvantage of clones mutant for the hypomorphic Rheb7A1 allele, further supporting the idea that Myc acts downstream of TOR to activate targets that control growth in these clones (Parisi, 2011).
The genetic interaction revealed a stronger dependence on Myc expression when Rheb was used as opposed to S6K. A possible explanation for this difference could lie in the fact that S6K is not capable of auto-activation of its kinase domain unless stimulated by TOR kinase. TOR activity is dependent on its upstream activator Rheb; consequently the enzymatic activity of the Rheb/GTPase is the limiting factor that influences S6K phosphorylation and therefore capable of maximizing its activity (Parisi, 2011).
Interestingly, these experiments also showed that activation of TOR signaling has a negative effect on the number of ommatidia, and this correlates with the ability of RhebAV4 to induce cell death during the development of the eye imaginal disc. Rheb-induced cell death was rescued in a dmP0 mutant background, which leading to the speculation that 'excessive' protein synthesis, triggered by overexpression of TOR signaling, could elicit a Myc-dependent stress response, which induces apoptosis. Alternatively, high protein synthesis could result in an enrichment of misfolded proteins that may result in a stress response and induces cell death. Further analysis is required to delineate the mechanisms underlying this process (Parisi, 2011).
This analyses provide novel genetic and biochemical evidences supporting a role for Myc in the integration of the insulin and TOR pathway during the control of growth, and highlights the role of GSK3β in this signaling. It was found that insulin signaling inactivates GSK3β to control Myc protein stability, and a similar biochemical regulation is also shared by activation of the TOR pathways. In support of this data, a recent genomic analysis in whole larvae showed a strong correlation between the targets of Myc and those of the TOR pathway; however, less overlap was found between the targets of Myc and those of PI3K signaling (Parisi, 2011).
Statistical analysis applied to the genetic interaction experiments revealed that, in the Drosophila eye, proliferation induced by activation of the insulin pathway is sensitive to variations in Myc levels, while a significant interaction was seen mostly when TOR increased cell size. The data therefore suggests that there is a correlation between Myc and the InR signaling and it is expected that the InR pathway also shares some transcriptional targets with Myc. Indeed, an overlap was found between the targets induced by insulin and Myc in Drosophila S2 cells and these targets have also been reported in transcriptome analyses in the fat body upon nutritional stress, suggesting that Myc acts downstream of InR/PI3K and TOR signaling and that this interaction might be specific to some tissues or in a particular metabolic state of the cell (Parisi, 2011).
The nutrient/target-of-rapamycin (TOR) pathway has emerged as a key regulator of tissue and organismal growth in metazoans. The signalling components of the nutrient/TOR pathway are well defined; however, the downstream effectors are less understood. This study shows that the control of RNA polymerase (Pol) III-dependent transcription is an essential target of TOR in Drosophila. TOR activity controls Pol III in growing larvae via inhibition of the repressor Maf1 and, in part, via the transcription factor Drosophila Myc (dMyc). Moreover, it was shown that loss of the Pol III factor, Brf, leads to reduced tissue and organismal growth and prevents TOR-induced cellular growth. TOR activity in the larval fat body, a tissue equivalent to vertebrate fat or liver, couples nutrition to insulin release from the brain. Accordingly, it was found that fat-specific loss of Brf phenocopies nutrient limitation and TOR inhibition, leading to decreased systemic insulin signalling and reduced organismal growth. Thus, stimulation of Pol III is a key downstream effector of TOR in the control of cellular and systemic growth (Marshall, 2012).
The TOR kinase is one of the best-established growth regulators. In virtually all animals, TOR activity can be stimulated by extracellular cues such as growth factors, nutrients and oxygen to control cell, tissue and organismal growth (Marshall, 2012).
Despite the knowledge of the signalling inputs to TOR, little is known about the mechanisms that allow TOR to modulate cell metabolism and drive growth. Most studies on metabolic functions modulated by TOR have been confined to yeast and mammalian cell culture. These studies have been important in defining roles for TOR in protein synthesis, nutrient uptake and metabolism and autophagy. But they leave open the question of what mechanisms operate in vivo to control tissue and organ growth during animal development. Genetic studies in Drosophila have been pivotal in this regard. This study shows that the ability of the TOR pathway to control transcription through Pol III governs cell, tissue and ultimately organismal growth in Drosophila. Given that Pol III drives transcription of several non-coding RNAs required for mRNA translation, it is suggested that the stimulation of Pol III by TOR enhances the protein synthetic capacity of cells. Previous study have shown that Drosophila TOR also controls synthesis of rRNA synthesis, via the RNA polymerase I factor, TIF-IA (Grewal, 2007
The Pol III transcription factor Brf has been shown to be an essential component of the TFIIIB complex responsible for recruiting Pol III to gene promoters. This work indicates that Brf activity is required for Drosophila development. Patterning and cell fate specification appear normal in brf embryos. However, once these mutants hatch as larvae they fail to grow. The data suggest that this growth arrest phenotype reflects a role for Brf activity downstream of TOR. Brf was found to be cell-autonomously required for growth in both endoreplicating cells, which make up the bulk of larval mass, and the mitotically dividing cells of the imaginal discs. In particular, brf mutant wing disc cell clones were found to be outcompeted by wild-type neighbours. This cell competition phenotype is seen in mutants for other genes required for protein synthesis, such as the ribosomal proteins and Myc. An important finding was that the overgrowth caused by loss of TSC1 (and hence increased TOR activity) was blocked in brf mutant cells. In mammalian cells, Brf activity is induced by cues that promote cell growth (e.g., during hypertrophic growth of cardiac cells) whereas cell differentiation leads to inhibition of Brf. In fact, overexpression of Brf alone can promote proliferation and transformation in immortalized fibroblasts. Mutations in tumour suppressors such as TSC are common in cancer and lead to elevated TOR activity and promotion of tumour growth. Based on the current data, it is suggested that Brf is required in vivo for both normal tissue growth and TOR-induced tumour growth (Marshall, 2012).
This study found that the predominant mechanism by which nutrition/TOR controls Pol III is via Maf1 repression, since Maf1 inhibition completely reverses the decrease in tRNA synthesis caused by reducing TOR activity. These findings extend those observed in both yeast and mammalian cell culture, and suggest an important role for dMaf1 in vivo in developing tissues. The exact mechanism by which Maf1 functions is not clear, but it may involve inhibition of Brf and Pol III recruitment to genes, possibly by direct binding or association with Brf/Pol III. Indeed, an enhanced association was seen between dMaf1 and Brf1 upon TOR inhibition. The role of dMyc was explored as a potential link between nutrient-TOR signalling and Pol III. dMyc was found to be both necessary and sufficient for the control of Pol III activity during development. As previously reported in both mammalian and Drosophila culture, it was possible to identify an interaction between dMyc and Brf (Gomez-Roman, 2003; Steiger, 2008). In addition, a role has been identified for dMyc in controlling the levels of components of the Pol III machinery, including both Trf and Brf which form part of the TFIIIB complex. Thus, dMyc likely has both direct and indirect effects on Pol III activity in Drosophila. These effects are necessary for both dMyc-induced cell growth (Steiger, 2008) and, as is shown in this study, for the non-autonomous increases in body size caused by dMyc in fat cells. Previous studies have shown that, in Drosophila, TOR controls Myc protein levels. But these effects on Myc probably do not play major role in how TOR activates Pol III since the data show that, unlike inhibition of Maf1, maintaining Myc levels and activity cannot reverse the decrease in tRNA synthesis caused by TOR inhibition. Moreover, if Myc protein levels were limiting for TOR-dependent control of Pol III, then it would not be expected that knockdown of Maf1 could completely reverse the effects of rapamycin/starvation. Given that Maf1 inhibition did not influence levels of Pol III factors, pre-rRNA or RP gene mRNA—transcripts that are upregulated by dMyc—it is unlikely that Maf1 influences Myc function. It was found that rapamycin feeding could not exacerbate the reduction of tRNA levels seen in dMyc null mutants. This result in principle may suggest that TOR signalling does not exert any dMyc-independent effects on Pol III function. But, it is suggested that this finding probably occurs because in the absence of Myc, Pol III activity may be approaching basal levels and cannot be significantly decreased much further. Taken together, although these data may not completely rule out some contribution of Myc to TOR-dependent control of Pol III, they do indicate that it is not the major contributor (Marshall, 2012).
It is clear that both TOR and Myc are essential regulators of Pol III. But, it is likely that while TOR can control Myc levels, both TOR and Myc can also function in parallel and independently of each other. Previous studies have shown that overactivation of TOR signalling could not promote growth when Myc was inhibited, but at the same time Myc overexpression could not promote growth when TOR was inhibited. These findings and the current data suggest that TOR and Myc cannot necessarily be placed in a simple, linear pathway. Recent studies in Drosophila have emphasized how other conserved growth-regulatory pathways, particularly those that control growth of the imaginal tissues (such as Wingless, EGF/Ras, the Hippo-Yorkie pathway and Bantam RNAi) function via control of dMyc. Thus, dMyc may play a role in coupling these pathways to the control of Pol III activity to stimulate cell growth and proliferation (Marshall, 2012).
It is interesting to speculate as to which Pol III targets are important for growth control. Pol III regulates the expression of several short non-coding RNAs, such as the tRNAs, 5S rRNA and 7SL RNA. Regulation of 5S rRNA production by Brf could influence ribosome synthesis and hence growth. However, it was found that loss of Brf did not inhibit Pol I activity or alter levels of rRNA, suggesting that Brf probably does not directly influence ribosome numbers. One attractive possibility is that levels of the tRNAs may be limiting for translation and growth. In support of this notion, a recent paper showed that overexpression of Brf increased tRNA levels and promoted proliferation and transformation of cultured mammalian fibroblasts (Marshall, 2008). These effects of Brf were phenocopied by just increasing levels of tRNAiMet, and were associated with augmented mRNA translation and increased protein levels of growth promoters such as c-Myc and cyclin D1. No consistent increase was seen in tRNAs when Brf was overexpressed in larvae, perhaps because levels of other components of the TFIIIB complex are limiting in flies. Nevertheless, by controlling Brf activity and tRNA synthesis, TOR could promote translation of growth regulators and drive larval growth. In fact, a recent paper (Teleman, 2008) indicated that TOR signalling in Drosophila regulates dMyc protein levels, but not dMyc mRNA levels, consistent with a possible role for translational control (Marshall, 2012).
One interesting result of this work was the identification of a non-cell autonomous role for Brf in organismal growth. Specifically, it was found that Brf activity in the fat cells of Drosophila larvae could influence larval growth and final size. A role for TOR in the fat body has been shown to exist as a relay to control peripheral insulin signalling. In feeding larvae, amino-acid input into fat cells activates TOR, leading to transmission of a secreted signal from fat to brain to increase dILP expression and release from brain IPCs. These data suggest that stimulation of Pol III activity may be an important downstream effector of this adipose function of TOR. Thus, adipose-specific silencing of Brf led to reduced peripheral insulin signalling, slower larval growth rate and reduced final body size. As in starved larvae, this study found that loss of brf led to reduced expression of dilp mRNA (seen in both brf mutants and cg>brf RNAi larvae) and reduced dILP release from the brain. Moreover, given that levels of phospho-Akt are lower, and levels of dInR (a FOXO target) are higher in tissues from both brf mutant and r4>brf RNAi larvae it is clear that systemic insulin signalling is reduced when Brf is inhibited in the fat body. This study also found that another fat phenotype associated with starvation and loss of TOR, accumulation of lipid droplets, was phenocopied by loss of Brf. However, the autophagy phenotype of starved larval fat bodies was not phenocopied by loss of Brf. Therefore, Brf and Pol III function in the Drosophila fat body may mediate some, but not all of TOR's effects on growth and metabolism. The exact nature of the fat-to-brain secreted factor that controls insulin release in flies is not yet known, but perhaps translation of this signal, if it is a peptide or secreted protein, is influenced by changes in tRNA synthesis and translation rates. Indeed, it has been shown that dMyc activity in the fat body was also important for controlling systemic insulin signalling, growth and body size. This effect of dMyc correlated with elevated expression of ribosome biogenesis genes and increased nucleolar size, an index of ribosome synthesis. dMyc overexpression can also stimulate Pol III and tRNA levels, and the increase in body size caused by fat body overexpression of dMyc is reversed by knockdown of Brf. These data suggest that regulation of mRNA translational capacity is a key step downstream of TOR and dMyc in fat cells to control signalling to IPCs (Marshall, 2012).
Together, these data suggest that mRNA translational control may underlie a role for the fat body as an endocrine organ. A similar theme is emerging in mouse models. Mammalian adipose tissue is known to secrete adipokines and leptin to influence organismal metabolism and growth. The secretion of many of these factors is influenced by diet, suggesting a regulatory role for TOR signalling. Genetic inhibition of either TOR and S6K in mice leads to alterations in metabolic activity in adipose tissue. Moreover, loss of the translational repressors, 4E-BP1 and 4E-BP2, both of which are downstream TOR effectors, alters lipid and glucose metabolism in mice. To date, there are no mouse models of Pol III. However, it is interesting to speculate that changes in Pol III and tRNA synthesis are involved in mediating effects of TOR in adipose tissue in mice. Regulation of Pol III by TOR may also be important in the metabolic control of other processes. For example, TOR is a conserved regulator of organismal stress responses and lifespan. These stress responses rely on TOR's ability to control translation. It is suggested that regulation of Pol III and tRNA synthesis may also be a mode of control. Further organismal studies, using genetic modulation of Pol III function, should provide additional insights into these points (Marshall, 2012).
PRAS40 has recently been identified as a protein that couples insulin/IGF signaling (IIS) to TORC1 activation in cell culture; however, the physiological function of PRAS40 is not known. This study investigate flies lacking PRAS40 (FlyBase Name: Lobe). Surprisingly, it was found, both biochemically and genetically, that PRAS40 couples IIS to TORC1 activation in a tissue-specific manner, regulating TORC1 activity in ovaries but not in other tissues of the animal. PRAS40 thereby regulates fertility but not growth of the fly, allowing distinct physiological functions of TORC1 to be uncoupled. The main function of PRAS40 in vivo is to regulate TORC1 activity, and not to act as a downstream target and effector of TORC1. Finally, this work sheds some light on the question of whether TORC1 activity is coupled to IIS in vivo (Pallares-Cartes, 2012).
PRAS40 has been proposed to link IIS to TORC1 in cell culture. Two reports showed that PRAS40 binds the TORC1 complex thereby inhibiting its activity, and that phosphorylation of PRAS40 by Akt relieves this inhibition (Nascimento, 2010; Sancak, 2007; Vander Haar, 2007). Other studies, however, identified PRAS40 as a TORC1 substrate, suggesting that the apparent inhibitory effects of PRAS40 on the canonical TORC1 substrates 4EBP and S6K may reflect competition for substrate binding. This would place PRAS40 downstream, rather than upstream of TORC1. Indeed, as these studies point out, PRAS40 might function concomitantly as a TORC1 substrate and a TORC1 regulator, regulating mTORC1 activity via direct inhibition of substrate binding. These studies have led to several open questions: (1) does PRAS40 regulate TORC1 activity in vivo, as it does in cell culture? (2) does PRAS40 link IIS to TOR activation in vivo? and (3) is the main function of PRAS40 to act as a TOR substrate or as a TOR regulator? These two options can be distinguished in an animal context. If the main function of PRAS40 is to regulate TORC1 activity (i.e., it is genetically upstream of TORC1), then PRAS40 mutant phenotypes should be rescued by reducing activity of TORC1 or of a TORC1 target other than PRAS40. If, instead, PRAS40 functions mainly as a TOR substrate downstream of TORC1, then loss of PRAS40 cannot be rescued by manipulating TORC1. No animal models for PRAS40 loss of function have yet been reported to address these questions (Pallares-Cartes, 2012).
One physiological function of IIS and TORC1 of particular relevance to this present study is the regulation of fertility. In Drosophila, insulin-like peptides (DILPs) secreted by neurosecretory cells regulate the rate of germline stem cell division in the ovary. This links metabolic status to fertility, so that rich nutrient conditions cause high DILP secretion, leading to increased egg production. If IIS is abrogated in the ovary, as in the case of chico or InR mutants, egg production is completely blocked and the animals are sterile. The defect in chico mutant ovaries is ovary-autonomous because transplantation of chico mutant ovaries into wild-type hosts, containing normal levels of DILPS, does not rescue their defects. At the cellular level, IIS and TORC1 regulate almost all aspects of oogenesis including the rate of proliferation of ovarian somatic and germline cells, germline stem cell maintenance, vitellogenesis, and oocyte loss. Interestingly, the roles of IIS and TORC1 in regulating fertility are highly conserved throughout evolution, regulating similar processes in Caenorhabditis elegans and in mammals. As in flies, reduction of IIS via knockout of IGF-1 or IRS-2 causes infertility in mice. As in flies, normal TORC1 in mice prevents oocyte loss (Thomson, 2010) and hyperactivation of IIS or TORC1 leads to premature activation of all primordial follicles, resulting in premature follicular depletion (Reddy, 2010; Sun, 2010). In sum, IIS and TORC1 play critical roles in regulating fertility in an evolutionarily conserved manner (Pallares-Cartes, 2012).
This study presents a PRAS40 loss-of-function animal model. By generating PRAS40 knockout Drosophila, the in vivo function of PRAS40, as well as the connection between IIS and TORC1, were studied. PRAS40 is shown function to link IIS to TORC1 in the animal. Unexpectedly, however, it does so in a tissue-specific manner, influencing TORC1 activity predominantly in the fly ovary, but not in other tissues of the animal. As a result, PRAS40 regulates development of the ovary, but not growth or proliferation of somatic tissues, thereby influencing animal fertility but not animal growth. Because PRAS40 is present in all tissues of the fly, this indicates PRAS40 is a link between IIS and TORC1 that can be switched on and off in a tissue-specific manner. Furthermore, PRAS40 knockout phenotypes can be rescued by inhibiting TORC1 or by reducing S6K gene dosage, indicating that PRAS40 functions mainly as a TORC1 inhibitor in vivo. Finally, this work sheds light on the conundrum whether the IIS and TORC1 signaling pathways are linked under normal physiological conditions, showing that they are indeed linked, but only in particular tissues (Pallares-Cartes, 2012).
Both biochemically and genetically this study found that PRAS40 and IIS do not affect TORC1 activity in most tissues during growth of the fly. Removal of PRAS40 does not cause elevated TORC1 activity in larvae and, in agreement with previous studies, removal of chico does not lead to reduced TORC1 activity in the adult body or in larvae. Removal of PRAS40 does not cause any size abnormalities in the fly, which is a very sensitive readout for TORC1 activity during development. It was surprising to find, however, that in ovaries both IIS and PRAS40 do affect TORC1 activity. TORC1 activity drops in ovaries of chico− animals, and increases in ovaries of PRAS40− animals. Furthermore, in ovaries, PRAS40 links IIS to TORC1 in that removal of both chico and PRAS40 leads to renormalized TORC1 activity. These biochemical data are reflected by genetic epistasis data. Chico mutant flies are completely infertile, laying no eggs, and this phenotype is rescued by removal of PRAS40. These data indicate that under normal physiological conditions, IIS activates TORC1 in a tissue-specific manner (Pallares-Cartes, 2012).
Does PRAS40 also link IIS to TORC1 in the male germline? The fact that mutation of PRAS40 rescues the infertility of PDK14/5 mutant males, and that PRAS40 mutant testes are larger than control testes suggests that it does. PRAS40−, chico− mutant testes also appear mildly increased in size compared to chico− mutant testes, however, the result is not as clear cut as with ovaries, because chico mutant females are completely sterile whereas chico mutant males have only mildly reduced fertility. Further work will be required to look at this carefully (Pallares-Cartes, 2012).
All these data, indicating an ovary-specific link between IIS and TORC1 result from manipulations within physiological range. In contrast, overexpression of PRAS40 does cause reduced tissue growth as well as reduced TORC1 activity, indicating that PRAS40 can inhibit TORC1 in most tissues when overexpressed. Furthermore, in contrast to the tissue-specific link between IIS and TORC1 under normal physiological conditions, it was also observed that hyper-stimulation of IIS above physiological range does activate TORC1 in most tissues, for instance in tissue explants treated with insulin, or in animals overexpressing activated PI3K (Dp110-CAAX). This mechanism might be relevant for pathophysiological conditions with elevated IIS, such as in cancer cells. This may occur via elevated ATP production in the cell, inhibiting AMPK, because this activation was also observed in tissues simultaneously lacking PRAS40 and all Akt phosphorylation sites on Tsc1 and Tsc2 (Pallares-Cartes, 2012).
One open question is whether the main function of PRAS40 is to regulate TORC1 activity or whether it functions mainly as a downstream target and effector of TORC1. The data suggest the former is the case. If PRAS40 had effector functions downstream of TORC1, these functions would not be rescued by additional removal of other TORC1 substrates such as S6K. Instead, it was found that the elevated fertility of PRAS40 mutants is rescued by removal of one copy of S6K, suggesting that the phenotype found in PRAS40 mutants is due to elevated S6K activity (Pallares-Cartes, 2012).
Why does PRAS40 regulate TORC1 activity in ovaries but not in other tissues of the animal? PRAS40 is expressed in all tissues that were tested. Therefore, the fact that removal of PRAS40 from larval tissues, for instance, has no effect on TORC1 activity must mean that larval PRAS40 protein is inactive. Data is presented suggesting that the state of phosphorylation of PRAS40 may be different in larval tissues compared to ovaries, providing a possible explanation for this inactivation. To date, a number of phosphorylations on PRAS40 have been reported, all of which are inhibitory in terms of TORC1 binding. These include phosphorylations by Akt, TORC1 itself, PIM1, and PKA. Intriguingly, this correlates with the observation that PRAS40 is highly phosphorylated in many cancers and that PRAS40 phosphorylation correlates with bad prognosis. The possibility is favored that PRAS40 phosphorylation on an inhibitory site could be regulated by a kinase that is absent in ovaries, or a phosphatase that is enriched in ovaries compared to other tissues. Future studies will shed light on this issue (Pallares-Cartes, 2012).
TORC1 has multiple physiological roles in various tissues. In Drosophila, TORC1 in the growing larva regulates both growth and metabolism of the animal whereas in the adult fly, it regulates mainly metabolic parameters. TORC1 in ovaries regulates fertility of the animal, whereas in the nervous system it regulates dendritic tiling. Therefore, unless TORC1 activity can be differentially regulated in various tissues, all these physiological functions would have to be controlled in a correlated fashion. Tissue-specific differential regulation of PRAS40 presents a mechanism that allows TORC1 activity to be uncoupled in a tissue-specific manner (Pallares-Cartes, 2012).
Endocycles are variant cell cycles comprised of DNA synthesis (S)- and gap (G)-phases but lacking mitosis. Such cycles facilitate post-mitotic growth in many invertebrate and plant cells, and are so ubiquitous that they may account for up to half the world's biomass. DNA replication in endocycling Drosophila cells is triggered by cyclin E/cyclin dependent kinase 2 (CYCE/CDK2), but this kinase must be inactivated during each G-phase to allow the assembly of pre-Replication Complexes (preRCs) for the next S-phase. How CYCE/CDK2 is periodically silenced to allow re-replication has not been established. This study used genetic tests in parallel with computational modelling to show that the endocycles of Drosophila are driven by a molecular oscillator in which the E2F1 transcription factor promotes CycE expression and S-phase initiation, S-phase then activates the PCNA/replication fork-associated E3 ubiquitin ligase CRL4CDT2 (Cul-4), and this in turn mediates the destruction of E2F1 (Shibutani, 2008). It is proposed that the transient loss of E2F1 during S phases creates the window of low Cdk activity required for preRC formation. In support of this model overexpressed E2F1 accelerated endocycling, whereas a stabilized variant of E2F1 blocked endocycling by deregulating target genes, including CycE, as well as Cdk1 and mitotic cyclins. Moreover, it was found that altering cell growth by changing nutrition or target of rapamycin (TOR) signalling impacts E2F1 translation, thereby making endocycle progression growth-dependent. Many of the regulatory interactions essential to this novel cell cycle oscillator are conserved in animals and plants, indicating that elements of this mechanism act in most growth-dependent cell cycles (Zielke, 2011).
Altogether these results indicate that periodic E2F1 degradation is necessary for endocycling for three reasons: (1) it creates a window of low CYCE/CDK2 activity; (2) it promotes high APCFzr/Cdh1 activity and thereby suppresses geminin accumulation; and (3) it allows E2F2 to maintain repression of CDK1 and its cyclins. Each of these conditions is required for preRC assembly and endocycle progression. This cell cycle mechanism is fundamentally different from that used in mitotic cycles, wherein destruction of the M-phase cyclins by APCCdc20/Fzy, rather than of E2F1 by the CRL4CDT2, throws the switch that allows preRC assembly. Indeed it is noteworthy that the periodic degradation of E2F1 and depletion of CYCE are not required for mitotic cell cycles in Drosophila. CRL4CDT2 is required for endocycling in plants, indicating that this element of the endocycle oscillator is conserved (Zielke, 2011).
Finally, it was asked what factors control E2F production to regulate endocycle rates. Endocycle speed and number can be manipulated by altering cell growth through changes in dietary protein or growth-regulatory genes including Myc and insulin/PI3K/TOR signalling components. Hence larvae were starved of protein to suppress insulin/TOR signalling, reduce protein synthesis, and block cell growth. Starvation arrested the salivary endocycles within 24h and strongly depleted E2F1. E2f1 and Dp mRNA levels were not affected, but the E2F targets CycE, pcna and rnrS were reduced. To test whether this was responsible for starvation-induced endocycle arrest E2F1 was overexpressed in the salivary glands of starved animals. Although these glands failed to grow their nuclei incorporated BrdU and accrued approximately sevenfold more DNA than controls. Overexpression of RHEB, which activates the Target of rapamycin (TOR) kinase and increases ribosome biogenesis and cap-dependent translation, also restored cell growth, E2F1 protein, and endocycle progression in starved animals. Thus E2F1 appears to act as a 'growth sensor' that couples rates of endocycle progression to rates of cell growth. A likely mechanism for this, corroborated by modelling, involves increased translation of E2F1 in rapidly growing cells. Indeed, it was found that the association of E2F1 mRNA with polyribosomes was greatly reduced in protein-starved animals. Translational control of E2F is an attractive mechanism for coupling growth to G1/S progression not only in endocycling cells, but also in growth-dependent mitotic cells with extended G1 periods (Zielke, 2011).
FoxO transcription factors, inhibited by insulin/insulin-like growth factor signalling (IIS), are crucial players in numerous organismal processes including lifespan. Using genomic tools, this study uncovered over 700 direct dFOXO targets in adult female Drosophila. dFOXO is directly required for transcription of several IIS components and interacting pathways, such as TOR, in the wild-type fly. The genomic locations occupied by dFOXO in adults are different from those observed in larvae or cultured cells. These locations remain unchanged upon activation by stresses or reduced IIS, but the binding is increased and additional targets activated upon genetic reduction in IIS. The part of the IIS transcriptional response directly controlled by dFOXO and the indirect effects were identified in this study, and it was show that parts of the transcriptional response to IIS reduction do not require dfoxo. Promoter analyses revealed GATA and other forkhead factors as candidate mediators of the indirect and dfoxo-independent effects. Genome-wide evolutionary conservation of dFOXO targets was identified between the fly and the worm Caenorhabditis elegans, enriched for a second tier of regulators including the dHR96/daf-12 nuclear hormone receptor (Alic, 2011).
Using ChIP-chip this study has defined >1400 genomic locations occupied by dFOXO in the adult fly. Interestingly, these locations are distinct from those observed by others in larvae. It is possible that the differences between the adult data and the published larval data stem from differences in protocols (e.g. the antibody used) or even experimental design (e.g. sex of the flies used). Importantly, however, this study showed that the observed differences between S2 cells and adults, in the case of the promoter (P1) and the coding region of the Drosophila InR, represent true biological differences. It is not surprising that dFOXO would occupy different locations during development and in the adult fly. A similar observation has been made for a number of transcriptional events, and even the dInR gene alone is transcribed from three promoters under tight spatio-temporal control. Furthermore, some differences will stem from cell- and tissue-specificity of dFOXO action. Indeed, FoxO factors are known to elicit tissue-specific transcriptional changes in the mouse, and the same tissue-restricted action by dFOXO on the transcription of the myc gene has been observed in Drosophila larvae. By binding to different locations in a spatially and temporally determined manner, dFOXO would be able to orchestrate different responses to suit its function in different life stages and tissues. Interestingly, a substantial portion of dFOXO bound in transcribed regions. In yeast, forkhead factors regulate Pol II elongation, and dFOXO may perform a similar function (Alic, 2011).
dFOXO was observed to be bound to a number of genes encoding IIS signalling components. Furthermore, dfoxo may also exert feedback onto other pathways that regulate it: dFOXO was bound near the genes encoding PP2A-B′, 14-3-3ε and JNKKKs (slpr and TAK1), among others. PP2A, 14-3-3ε and JNK have all been shown to regulate FoxO activity. A number of these dFOXO-activated genes is also activated on over-expression of superoxide dismutase, suggesting that dFOXO, like its mammalian counterparts, may be redox regulated. Interestingly, binding was detected to only the intracellular components of IIS such as chico, Lnk and Akt, while the genes with altered expression level in dfoxo-/- include extracellular cell-to-cell signalling molecules, such as those encoded by dilp3, dilp6 and Imp-L2. The latter genes have a more localised expression pattern, for example dilp3 is expressed in only ~14 cells in the whole adult fly. It is possible that genes such as dilp3 are also bound and directly regulated by dFOXO but that this was not observed in the whole fly ChIP-chip due to a very small number of cells in which this binding occurs (Alic, 2011).
4E-BP (a.k.a. Thor) has been shown to be bound by dFOXO in larvae, and its regulation has been reported as consistent with dFOXO acting as a direct activator of its expression. On the other hand, dFOXO binding was not observed in the vicinity of this gene in adults, and the 4E-BP transcript is actually elevated in a dfoxo null. It is possible that dFOXO is required for direct activation of this gene in only a limited number of cells/tissues in the adult, thus escaping detection by ChIP-chip on whole animals. Furthermore, the role of dFOXO in 4E-BP regulation may be sexually dimorphic. Alternatively, 4E-BP might be a target of a different forkhead factor in the adult female fly. Indeed, Forkhead (Fkh, the fly FoxA orthologue) is able to activate transcription of 4E-BP in larvae. Since dfoxo nulls have reduced levels of TOR, and TOR is an inhibitor of Fkh activity, it is likely that Fkh is activated in dfoxo nulls leading to increased levels of the 4E-BP transcript. It remains to be established whether Fkh might indeed be directly binding to the 4E-BP locus in adult flies (Alic, 2011).
From the 1400 dFOXO-bound locations, using transcriptional profiling of dfoxo null flies under normal conditions or with reduced IIS, >700 direct transcriptional targets of dFOXO were identified in the adult. Several functions associated with these genes have been linked with FoxO biology previously, such as cell cycle, cytoskeleton organisation, negative regulation of gene expression such as translation and regulation of protein catabolism. dFOXO is known to be involved in the repression of protein synthetic machinery via myc in larvae but this study also revealed a significant regulation of ribosome biogenesis genes effected directly by dFOXO in the adult female. Other, previously unknown functions were identified, such as control of negative regulators of transcription and chromatin modifiers, hinting at the importance of dFOXO in establishment and maintenance of repressive chromatin states. Yet other functions were completely unexpected. For example, dFOXO appears as a positive regulator of sexual reproduction, including oogenesis, in an IIS mutant. This surprising finding is backed up by phenotypic epistasis analysis that shows removal of dfoxo to exacerbate the fecundity defect of several IIS mutants. Hence, dFOXO actually positively regulates some aspects of IIS. Indeed, one of the most surprising findings of this study is that dFOXO is directly required for expression of several components of IIS and interacting pathways, including TOR and Sos, in the wild-type fly, with consequences for the downstream signalling events. Importantly, this is not just simple feedback in response to alteration in the levels of insulin/IGF-like signal, but rather dFOXO is active in the normal adult and its activity promotes signalling through the IIS pathway. This observation can also explain why dfoxo deletion is lethal in combination with certain IIS mutants, since the combined reduction in IIS will be too great for the flies to survive. This potentiation of IIS by FoxOs could also explain why mice with reduced IIS through mutation of IRS1 have mild insulin resistance but preserved old-age glucose homoeostasis. In this case, the mild insulin resistance would be the primary effect of the mutation of IRS1, while the resulting activation of FoxOs would be responsible for sustained IIS in old age and thus for the observed preservation of glucose homoeostasis (Alic, 2011).
dFOXO directly regulates an extensive second tier of regulators; throughout this study different transcriptional and post-transcriptional regulators were repeatedly encountered as predominant dFOXO targets. This aspect of dFOXO biology is also conserved in the worm. Indeed, some of the potential secondary effectors are directly conserved between the worm and the fly, such as the nuclear hormone receptor dHR96/daf-12, highlighting their importance. This study also illustrates the role this second tier of regulators may play. dFOXO is directly required for the maintenance of GATAd mRNA levels in both the wild-type and IIS-compromised flies, and this in effect may constitute an IIS feed-forward loop, since GATAd in turn may be an important transcriptional repressor in response to reduced IIS. Hopefully, subsequent studies will demonstrate the existence of such a feed-forward loop (Alic, 2011).
Since daf-16 is strictly required for all phenotypic outputs of reduced IIS in the worm, and also appears strictly required for the transcriptional response to reduced IIS, it was very surprising to find that dFOXO was only required for part of the transcriptional response to reduced IIS in the fly. On the other hand, this is in accordance with phenotypic epistasis experiments in the fly where lifespan extension and xenobiotic resistance are dependent on dfoxo, while lowered fecundity and body size, delayed development and resistance to paraquat are not. This implies that phenotypes such as fecundity are negatively regulated via other factors in the fly. This study indicates that GATA factors are the most likely candidates for mediating transcriptional repression in response to reduced IIS. Studies in the worm have also revealed the presence of a GATA-recognition sequence in the promoters of IIS-regulated genes. Furthermore, at least one of the 14 worm GATA TF (elt-3) is regulated by IIS, and reduced function in any of the three GATA TFs (elt-3, egr-1, egl-27) blocks the lifespan extension by a daf-2 mutant. The role of GATA factors in lifespan in other organisms awaits examination. At the same time, this study reveals the potential involvement of other forkhead factors, besides dFOXO, in the transcriptional activation response to IIS reduction. Fkh is the prime suspect, since it is regulated by TOR signalling in the fly, and Foxa2 is involved in the IIS response in mammals. Indeed, Foxa2 is directly inactivated by Akt via phosphorylation of a single site that is conserved in the fly Fkh. While this study provides hints, further work will be needed to determine the identity of other TFs involved in the fly IIS response (Alic, 2011).
This study reveals that the transcriptional response to IIS in the fly is clearly more complex than that in the worm. The parallel genetic study performed by (Slack (2011) shows that the genes directly regulated by dFOXO must still effect the lifespan extension by reduction in IIS. Importantly, this study has now identified these genes. Their characterisation is the next step towards understanding the physiological and molecular changes that can extend animal lifespan, keeping in mind that it is now crucial to determine the architecture of the mammalian response to reduced IIS (Alic, 2011).
Loss-of-function mutations in TRPML1 (transient receptor potential mucolipin 1) cause the lysosomal storage disorder, mucolipidosis type IV (MLIV). This study reports that flies lacking the TRPML1 homolog display incomplete autophagy and reduced viability during the pupal period -- a phase when animals rely on autophagy for nutrients. TRPML is required for fusion of amphisomes with lysosomes, and its absence leads to accumulation of vesicles of significantly larger volume and higher luminal Ca2+. It was also found that trpml1 mutant cells showed decreased TORC1 (target of rapamycin complex 1) signaling and a concomitant upregulation of autophagy induction. Both of these defects in the mutants are reversed by genetically activating TORC1 or by feeding the larvae a high-protein diet. The high-protein diet also reduces the pupal lethality and the increased volume of acidic vesicles. Conversely, further inhibition of TORC1 activity by rapamycin exacerbates the mutant phenotypes. Finally, TORC1 exerts reciprocal control on TRPML function. A high-protein diet causes cortical localization of TRPML, and this effect is blocked by rapamycin. These findings delineate the interrelationship between the TRPML and TORC1 pathways and raise the intriguing possibility that a high-protein diet might reduce the severity of MLIV (Wong, 2012).
Drosophila transient receptor potential mucolipin (TRPML) localizes to late endosomes (LEs)/lysosomes in cultured cells (Venkatachalam, 2008). To identify the subcellular localization of TRPML in vivo, a UAS-trpml::myc transgene was expressed in flies using the GAL4/UAS system. TRPML::MYC decorated the periphery of LysoTracker-positive vesicles and colocalized with the LE/lysosomal markers YFP::Rab7 and lysosome-associated membrane protein:green fluorescent protein (LAMP::GFP). These data indicate that TRPML::MYC is a LE/lysosomal membrane protein, as is the case for mammalian TRPML1 (Venkatachalam, 2006; Wong, 2012 and references therein).
The trpml1 flies are unable to complete lysosomal degradation of autophagosomes. To identify the step in the lysosomal degradation pathway affected in trpml1, the degradation of the Drosophila Wnt homolog, Wingless (Wg) was evaluated. Following binding to its receptor, Wg is internalized into endosomes and degraded in lysosomes. It was found that there was increased accumulation of Wg in the wing pouch and notum of trpml1 wing discs, and this phenotype was rescued by a trpml+ genomic transgene (P[trpml+];trpml1) (Wong, 2012).
Wg could be accumulating either in early endosomes or in LEs/multivesicular bodies (MVBs) in trpml1 discs. To discriminate between these possibilities, the observation was taken into account that Wg transmits signals at both the plasma membrane (PM) and early endosomes. Only after the formation of MVBs is the signal terminated. Therefore, increased Wg signaling in trpml1 would suggest that Wg is accumulating in early endosomes, whereas unchanged Wg signaling would be consistent with Wg accumulating in MVBs. Therefore, activation of the Wg target gene Hindsight (Hnt) in wing discs was evaluated. Nuclear Hnt expression was indistinguishable between wild-type (WT) and trpml1, indicating that Wg signaling was not increased in trpml1 (Wong, 2012).
Accumulation of Notch was also evaluated using an antibody specific for the endocytosed domain of Notch, and it was found that levels of Notch increased dramatically in trpml1 wing discs. Notch levels appeared higher than Wg in trpml1 because whereas Wg accumulated in the wing pouch and notum, Notch was elevated over the whole disc. In support of the conclusion that the vesicles that accumulated in trpml1 were LEs, the Notch-positive vesicles costained with LysoTracker (Wong, 2012).
Autophagy is a pathway required for the degradation of cellular macromolecules that are too big to fit through the proteosomal barrel. During autophagy, double-membrane-bound vesicles called autophagosomes isolate the cytosolic material destined for degradation. Subsequently, autophagosomes fuse with LEs/MVBs to form amphisomes. Amphisomes then coalesce with lysosomes leading to the formation of autolysosomes. Because lysosomes carry degradatory enzymes, the contents of amphisomes are broken down following autolysosome formation (Wong, 2012).
It has previously been reported that trpml1 adults display hallmarks of decreased autophagic flux (Venkatachalam, 2008). To provide evidence that there was accumulation of autophagosome and amphisomes, WT and trpml1 fat bodies were stained with LysoTracker and GFP::ATG8. Autophagosomes are labeled with GFP::ATG8 only, whereas amphisomes are stained with both GFP::ATG8 and LysoTracker. Although WT showed virtually no GFP::ATG8 staining, there were many trpml1 vesicles that were labeled with GFP::ATG8 only or both GFP::ATG8 and LysoTracker. These data indicated that loss of trpml led to an elevation of autophagosomes and amphisomes. Defects in receptor degradation have also been reported in human cells lacking TRPML1 and in C. elegans with a mutation disrupting the worm TRPML1 homolog. These trpml1 phenotypes closely resemble those of flies lacking fab1 (vesicular phosphatidylinositol 3-phosphate 5-kinase). These phenotypic similarities are consistent with the finding that the mammalian TRPML1 is activated by the product of Fab1/PIK-FYVE-kinase, PI(3,5)P(2) (Wong, 2012).
The sizes of LysoTracker-positive vesicles were compared in WT and trpml1 larval fat bodies to assess autophagy induction. Consistent with increased induction of autophagy in trpml1, there was a striking elevation in the volume of LysoTracker-positive vesicles in mutant fat bodies. This change became evident and was most pronounced in fat bodies from second-instar larvae. The difference between WT and trpml1 was less pronounced but still significant in third-instar larvae. The smaller elevation in trpml1 third-instar larvae likely reflects ecdysone-dependent autophagy activation in WT tissues at this developmental stage (Wong, 2012).
It was hypothesized that induction of autophagy without its completion should suppress target of rapamycin complex 1 (TORC1) activity due to two factors. First, a decline in autophagic flux would decrease net availability of amino acids that are produced via autophagic degradation of proteins. Reduced amino acid levels would diminish activity of the TORC1. Indeed, diminished autophagic flux by Atg7 knockdown led to reduced TORC1 activity as determined by phosphorylation of the TORC1 substrate, S6 kinase (pS6K). This study found a similar decrease in TORC1 activity after knocking down Atg5 in WT fat bodies using RNAi. Second, increased autophagy will directly suppress TORC1 function because autophagy and TORC1 activity are mutually antagonistic. Decreased TORC1 will induce further induction of autophagy leading to the generation of larger LysoTracker-positive vesicles (Wong, 2012).
Several lines of evidence support the preceding proposal. First, feeding trpml1 third-instar larvae protein-rich yeast paste suppressed the increase in the volume of LysoTracker-positive vesicles. Second, pS6K was diminished in trpml1 fat bodies. The decrease in pS6K in trpml1 was reversed by driving WT trpml+ in fat bodies using cg-GAL4. Furthermore, yeast feeding suppressed the reduction in pS6K levels in trpml1. To investigate whether decreased TORC1 activity occurs in other mutants with diminished fusion of LEs with lysosomes, pS6K was evaluated in dor mutants. Extracts from dor4 mutant larvae showed a reduction. Therefore, a block in the fusion of LEs with lysosomes results in decreased cellular amino acid levels and decreased TORC1 activity (Wong, 2012).
Third, genetically upregulating TORC1 activity in mutant fat bodies by overexpressing Rheb and constitutively active Rag (RagQ61L) (Kim, 2008) decreased the LysoTracker-positive vesicular volume. Vesicular volume in trpml1 did not increase any further when dominant-negative Rag (RagT16N) was expressed, indicating that Rag activity was already maximally reduced in trpml1. The finding that elevating TORC1 activity is sufficient to suppress lysosomal storage argues that the increase in acidic vesicles in trpml1 reflects a decrease in TORC1 activity. Therefore, elevating TORC1 activity is sufficient to prevent vesicle accumulation despite the persistence of vesicle fusion defects (Wong, 2012).
Finally, the half-maximal time to pupation was increased in trpml1. Because decreased TORC1 activity causes a developmental delay, these data are also consistent with a decrease in TORC1 activity in trpml1. Feeding the larvae protein-rich yeast paste restored pS6K levels to WT and rescued the defect in developmental timing (Wong, 2012).
Late endosome/lysosomal Ca2+ is required for the homotypic and heterotypic fusion of these vesicles. Therefore, the increased LysoTracker staining in trpml1 may have resulted from diminished Ca2+ release from the vesicles leading to impaired fusion of the early endosome/amphisomes with lysosomes, ultimately resulting in reduced degradation of their contents. Consistent with the proposal that increased vesicular volume stemmed from diminished Ca2+ release, treatment of the fat bodies with thapsigargin, which blocks the sarcoplasmic/endoplasmic reticulum calcium ATPase pump and causes Ca2+ release from endoplasmic reticulum stores, resulted in a significant decrease in the volume of LysoTracker-positive vesicles in trpml1. Therefore, despite the absence of a late endosome Ca2+ release mechanism in trpml1, elevation of cytosolic Ca2+ levels from a different Ca2+ reserve was sufficient to partially suppress the increase in LysoTracker-positive vesicle volume. Although the data are most consistent with a defect in the fusion of vesicles in trpml1, it cannot be ruled out that there may also be a defect in vesicular trafficking, thereby reducing encounters between fusible vesicles (Wong, 2012).
During the pupal period Drosophila do not feed, and they depend on autophagy for the amino acids necessary for morphogenesis and survival. Loss of trpml causes semilethality during the pupal period because <10% of adults eclose from the pupal cases (Venkatachalam, 2008). To test whether this reduced viability resulted from an insufficient supply of amino acids, the mutant larvae were fed a high-protein diet (food supplemented with 20% w/v yeast). It was found that this diet significantly suppressed the lethality. However, the effects of another mutation that caused pupal lethality (P element inserted in vamp-7, CG1599EY09354) were not suppressed by yeast supplementation (Wong, 2012).
The suppression of the trpml semilethality by yeast paste could have been due to either protein or carbohydrates in this supplement. Therefore, whether supplementation of either tryptone or sucrose diminished the lethality was tested. It was found that whereas tryptone supplementation reduced the semilethality, sucrose supplementation did not. The lack of suppression with sucrose indicated that the phenotype was not a result of caloric deprivation but rather reflected a requirement for increased dietary amino acids by trpml1 larvae (Wong, 2012).
Next, whether the suppression of the semilethality by the high-protein diet was due to increased TORC1 activity was considered. Yeast paste was fed to trpml1 larvae in the presence of the TORC1 inhibitor, rapamycin. It was found that rapamycin prevented suppression of the pupal semilethality by yeast paste. Moreover, rapamycin enhanced the lethality when trpml1 larvae were reared on normal food. However, feeding the dor mutants rapamycin did not decrease their viability. These data indicate that not all mutants with deficient fusion of LEs to lysosomes show increased sensitivity to rapamycin (Wong, 2012).
TORC1 simultaneously increases protein translation and decreases autophagy. One of the ways through which TORC1 increases protein translation is phosphorylation and inhibition of the translational suppressor Thor -- fly homolog of 4E-BP1. Therefore, if the effects of TORC1 activation in trpml1 depend upon protein translation, then rapamycin should not reverse the beneficial effect of yeast feeding in thor2;trpml1 double-mutant animals. However, yeast paste still suppressed the semilethality in thor2;trpml1 double mutants (no thor2;trpml1 adults eclosed in the absence of yeast-supplemented diet), and the effect of rapamycin remained unchanged. These results suggest that activation of TORC1 by high levels of amino acids may have suppressed the pupal semilethality of trpml1 by decreasing autophagy rather than by increasing protein translation (Wong, 2012).
To investigate whether TORC1 activity reciprocally affected TRPML, the spatial distribution was examined of TRPML::MYC under conditions in which TORC1 activity was manipulated. On a normal diet, TRPML::MYC was exclusively intracellular. However, on a high-protein diet, TRPML::MYC colocalized with the cortical F-actin marker phalloidin, indicating that TRPML was at the plasma membrane. In larvae maintained on a high-protein diet and rapamycin, TRPML::MYC was detected exclusively in intracellular vesicles. These data indicate that the PM localization of TRPML::MYC depends on the activity of TORC1. Further supporting this conclusion, TRPML::MYC was predominantly localized to the PM in larval salivary glands, which are characterized by low levels of autophagy (and therefore high TORC1 activity) until the onset of the pupal phase, when autophagy is required for the degradation of the pupal salivary gland during metamorphosis (Wong, 2012).
The effect of TORC1 activity on the subcellular location of TRPML is unlikely to reflect alterations in bulk endocytosis because TORC1 enhances rather than suppresses bulk endocytosis. Rather, it is suggested that by occluding entry of TRPML into endosomes and diminishing the levels of TRPML in the late endosomes, TORC1 exerts feedback regulation on the completion of autophagy. Therefore, in addition to suppressing the initiation of autophagy, TORC1 also inhibits the completion of autophagy by regulating the subcellular location of TRPML (Wong, 2012).
TRPML is a Ca2+ channel, which is endocytosed from the PM and eventually enters the late endosomes (LEs). The LEs fuse with autophagosomes creating amphisomes. TRPML present in amphisomes releases luminal Ca2+ to facilitate Ca2+-dependent fusion of amphisomes with lysosomes. The amino acids generated by degradation of proteins in the autolysosomes promote TORC1 activation. In addition to inhibiting the initiation of autophagy, activated TORC1 also diminishes endocytosis of TRPML (Wong, 2012).
In the absence of TRPML, fusion of amphisomes and lysosomes is impaired. This leads to a decrease in autophagic flux of amino acids causing a reduction in TORC1 and upregulation of autophagy. Supplementing the trpml1 diet with protein-rich yeast reverses the effects of diminished TORC1 activity. These findings raise the possibility that patients with mucolipidosis type IV (MLIV) may also show diminished TORC1 activity. If so, it is intriguing to speculate that amino acid supplementation might reduce the severity of the clinical manifestations associated with MLIV (Wong, 2012).
Neuronal differentiation is exquisitely controlled both spatially and temporally during nervous system development. Defects in the spatiotemporal control of neurogenesis cause incorrect formation of neural networks and lead to neurological disorders such as epilepsy and autism. The mTOR kinase integrates signals from mitogens, nutrients and energy levels to regulate growth, autophagy and metabolism. The insulin receptor (InR)/mTOR pathway has been identified as a critical regulator of the timing of neuronal differentiation in the Drosophila melanogaster eye. This pathway has also been shown to play a conserved role in regulating neurogenesis in vertebrates. However, the factors that mediate the neurogenic role of this pathway are completely unknown. To identify downstream effectors of the InR/mTOR pathway transcriptional targets of mTOR were screened for neuronal differentiation phenotypes in photoreceptor neurons. The conserved gene unkempt (unk), which encodes a zinc finger/RING domain containing protein, as a negative regulator of the timing of photoreceptor differentiation. Loss of unk phenocopies InR/mTOR pathway activation and unk acts downstream of this pathway to regulate neurogenesis. In contrast to InR/mTOR signalling, unk does not regulate growth. unk therefore uncouples the role of the InR/mTOR pathway in neurogenesis from its role in growth control. The gene headcase (hdc) was identified a second downstream regulator of the InR/mTOR pathway controlling the timing of neurogenesis. Unk forms a complex with Hdc, and Hdc expression is regulated by unk and InR/mTOR signalling. Co-overexpression of unk and hdc completely suppresses the precocious neuronal differentiation phenotype caused by loss of Tsc1. Thus, Unk and Hdc are the first neurogenic components of the InR/mTOR pathway to be identified. Finally, Unkempt-like is expressed in the developing mouse retina and in neural stem/progenitor cells, suggesting that the role of Unk in neurogenesis may be conserved in mammals (Avet-Rochex, 2014).
Neural progenitors in the developing human brain generate up to 250,000 neurons per minute. After differentiating from these neural progenitors, neurons migrate and are then integrated into neural circuits. Temporal control of neurogenesis is therefore critical to produce a complete and fully functional nervous system. Loss of the precise temporal control of neuronal cell fate can lead to defects in cognitive development and to neurodevelopmental disorders such as epilepsy and autism (Avet-Rochex, 2014).
Mechanistic target of rapamycin (mTOR) signalling has recently emerged as a key regulator of neurogenesis. mTOR is a large serine/threonine kinase that forms two complexes, known as mTORC1 and mTORC2. mTORC1 is rapamycin sensitive and is regulated upstream by mitogen signalling, such as the insulin receptor (InR)/insulin like growth factor (IGF) pathway, amino acids, hypoxia, cellular stress and energy levels. mTORC1 positively regulates a large number of cellular processes including growth, autophagy, mitochondrial biogenesis and lipid biosynthesis and activation of mTOR has been linked to cancer. Hyperactivation of mTOR signalling in neurological disease is best understood in the dominant genetic disorder tuberous sclerosis complex (TSC), which causes epilepsy and autism. mTOR signalling has also been shown to be activated in animal models of epilepsy and in human cortical dysplasia (Avet-Rochex, 2014).
The control of neurogenesis by the InR/mTOR pathway was first discovered in the developing Drosophila melanogaster retina, where activation of the pathway caused precocious differentiation of photoreceptor neurons and inhibition caused delayed differentiation. Subsequent in vitro studies demonstrated that insulin induces neurogenesis of neonatal telencephalonic neural precursor cells in an mTOR dependent manner and that Pten negatively regulates neuronal differentiation of embryonic olfactory bulb precursor cells. More recently, in vivo studies have shown that inhibition of mTOR suppresses neuronal differentiation in the developing neural tube. Furthermore, knock-down of the mTOR pathway negative regulator RTP801/REDD1 causes precocious differentiation of neural progenitors in the mouse embryonic subventricular zone (SVZ), while overexpression of RTP801/REDD1 delays neuronal differentiation. Loss of Pten, Tsc1, or overexpression of an activated form of Rheb, also cause premature differentiation of neurons in the SVZ. These studies have demonstrated that InR/mTOR signalling plays a conserved role in regulating neurogenesis in several different neural tissues. However, the downstream effectors of InR/mTOR signalling in neurogenesis are completely unknown (Avet-Rochex, 2014 and references therein).
To identify neurogenic downstream regulators of InR/mTOR signalling, genes were screened that were previously shown to be transcriptionally regulated by mTOR in tissue culture cells, for in vivo neurogenic phenotypes in the developing Drosophila retina. From this screen the zinc finger/RING domain protein Unkempt (Unk) was identified as a negative regulator of photoreceptor differentiation. Loss of unk phenocopies the differentiation phenotype of InR/mTOR pathway activation and Unk expression is negatively regulated by InR/mTOR signalling. Importantly, unk does not regulate cell proliferation or cell size and so uncouples the function of InR/mTOR signalling in growth from its role in neurogenesis. The evolutionarily conserved basic protein Headcase (Hdc) was identified as a physical interactor of Unk, and it was shown that loss of hdc causes precocious differentiation of photoreceptors. Hdc expression is regulated by the InR/mTOR pathway and by unk, demonstrating that Hdc and Unk work together downstream of InR/mTOR signalling in neurogenesis. Unk also regulates the expression of and interacts with D-Pax2 (Shaven/Sparkling), suggesting a model for the regulation of neurogenesis by the InR/mTOR pathway. It was also shown that one of the mammalian homologs of Unk, Unkempt-like, is expressed in the developing mouse retina and in the early postnatal brain. This study has thus identified the Unk/Hdc complex as the first component of the InR/mTOR pathway that regulates the timing of neuronal differentiation (Avet-Rochex, 2014).
Several lines of evidence together demonstrate that unk and hdc act downstream of InR/mTOR signalling to negatively regulate the timing of photoreceptor cell fate. First, loss of either unk or hdc causes precocious differentiation of the same cells and to the same degree as activation of InR/mTOR signalling. Second, the expression of both Unk and Hdc are regulated by InR/mTOR signalling. Third, loss of unk suppresses the strong delay in photoreceptor differentiation caused by inhibition of the InR/mTOR pathway and combined overexpression of unk and hdc suppresses the precocious photoreceptor differentiation caused by loss of Tsc1. Fourth, although Unk has been shown to physically interact with mTOR, neither unk nor hdc regulate cell or tissue growth. Taken together these data show that unk and hdc are novel downstream components of the InR/mTOR pathway that regulate the timing of neuronal differentiation (Avet-Rochex, 2014).
InR/mTOR signalling is a major regulator of cell growth. In Drosophila activation of InR/mTOR signalling by loss of either Tsc1, Tsc2, Pten, or overexpression of Rheb causes increased cell size and proliferation. In the genetic disease TSC, which is caused by mutations in Tsc1 or Tsc2, patients develop benign tumours in multiple organs including the brain. The previously identified components of the InR/mTOR pathway regulate both growth and neurogenesis in Drosophila and vertebrate model. unk and hdc therefore represent a branchpoint in the pathway where its function in neurogenesis bifurcates from that in growth control. Moreover, analysis of unk and hdc demonstrates that regulation of cell growth can be uncoupled from and is not required for the function of InR/mTOR signalling in the temporal control of neuronal differentiation (Avet-Rochex, 2014).
At the protein level this study shows that Unk and Hdc physically interact in S2 cells. Although this interaction remains to be demonstrated in vivo, the additional observations that they both regulate each other's expression and act synergistically in vivo strongly support the model that they physically interact (see A model for the regulation of the timing of neuronal differentiation by the Unk/Hdc complex acting downstream of InR/mTOR signalling). Moreover, Unk and Hdc have also previously been shown to physically interact by yeast-2-hybrid and co-immunoprecipitation. Unk and Hdc are both expressed in all developing photoreceptors and so it is hypothesised that they control the timing of differentiation through the regulation of neurogenic factors whose expression is restricted to R1/6/7 and cone cells. Loss of unk causes increased expression of D-Pax2, the main regulator of cone cell differentiation. hdc and Tsc1 mutant clones also cause a similar increase in D-Pax2 expression. Overexpression of D-Pax2 alone is insufficient to induce cone cell differentiation, which requires overexpression of both D-Pax2 and Tramtrack88 (TTK88). Thus, regulation of D-Pax2 expression by mTOR signalling may contribute to the rate of cone cell differentiation, while overall control would require the regulation of additional factors such as TTK88. Pax8, part of the Pax2/Pax5/Pax8 paired domain transcription factor subgroup that is homologous to D-Pax2, has been shown to physically interact with one of the two human homologs of Unkempt. This study found that Drosophila Unk physically interacts with D-Pax2, demonstrating that the physical interaction between Unk and this group of transcription factors is conserved. It is suggested that D-Pax2 may be one of several neurogenic factors regulated by InR/mTOR signalling, through a physical interaction with the Unk/Hdc complex, to control the timing of R1/6/7 and cone cell fate (Avet-Rochex, 2014).
Unk has been shown to physically interact with mTOR and the strength of this interaction is regulated by insulin. This suggests the intriguing possibility that the inhibition of Unk activity by InR/mTOR signalling is dependent on the strength of the physical interaction between Unk and the mTORC1 complex. Unk was also identified as part of the mTOR-regulated phosphoproteome in both human and murine cells. Thus, Unk may potentially be regulated by mTOR through phosphorylation. Future studies will fully characterise the mechanism by which mTORC1 regulates Unk activity (Avet-Rochex, 2014).
This study represents the first demonstration of a role for unk in specific developmental processes. By contrast, hdc has previously been shown to regulate dendritic pruning during metamorphosis and to act as a branching inhibitor during tracheal developmen. A screen for genes affecting tracheal tube morphogenesis and branching recently identified Tsc1, suggesting that InR/mTOR also regulates tracheal development. Thus, hdc and unk may act repeatedly as downstream effectors of the InR/mTOR pathway during Drosophila development (Avet-Rochex, 2014).
The one previous study of either of the mammalian Unk homologs showed that Unkl binds specifically to an activated form of the Rac1 GTPase. If this function is conserved in Drosophila then the defects in photoreceptor apical membrane morphogenesis caused by activation of mTOR signalling or loss of unk/hdc may be mediated through Rac1 (Avet-Rochex, 2014).
The function of the two unk homologs, unk and unkl, in mammalian development is not known, but unk has been shown to be expressed in the mouse early postnatal mouse retina. This study found that Unkl is also expressed in the developing mouse retina, suggesting that Unk may play a conserved role in eye development in both flies and mammals. InR/mTOR signalling acts as a pro-survival pathway preventing retinal degeneration, but its role in mammalian eye development has not been characterised. By contrast InR/mTOR signalling has a well characterised role in NSC self-renewal and differentiation in the mouse SVZ. Loss of Tsc1 or expression of a constitutively active form of Rheb in neural progenitor cells in the postnatal mouse SVZ causes the formation of heterotopias, ectopic neurons and olfactory micronodules. Furthermore, individuals with TSC, which results in activated mTOR signalling, have aberrant cortical neurogenesis and develop benign cortical tumours during foetal development and throughout childhood. mTOR signalling has been shown to be active in proliferative NSCs and TAPs in the neonatal SVZ and inhibition of mTOR signalling prevents NSC differentiation. This study found that Unkl is expressed in both NSCs and TAPs in the early postnatal SVZ. Thus, Unkl may regulate NSC differentiation downstream of mTOR signalling in the mammalian brain. Unkempt may therefore play a conserved role in regulating the timing of neural cell fate downstream of mTOR signalling in both flies and mammals (Avet-Rochex, 2014).
The v-ATPase is a fundamental eukaryotic enzyme that is central to cellular homeostasis. Although its impact on key metabolic regulators such as TORC1 is well documented, knowledge of mechanisms that regulate v-ATPase activity is limited. This study reports that the Drosophila transcription factor Mitf is a master regulator of the v-ATPase holoenzyme. Mitf directly controls transcription of all 15 v-ATPase components through M-box cis-sites and this coordinated regulation affects holoenzyme activity in vivo. In addition, through the v-ATPase, Mitf promotes the activity of TORC1, which in turn negatively regulates Mitf. Evidence is provided that Mitf, v-ATPase and TORC1 form a negative regulatory loop that maintains each of these important metabolic regulators in relative balance. Interestingly, direct regulation of v-ATPase genes by human MITF also occurs in cells of the melanocytic lineage, showing mechanistic conservation in the regulation of the v-ATPase by MITF family proteins in fly and mammals. Collectively, this evidence points to an ancient module comprising Mitf, v-ATPase and TORC1 that serves as a dynamic modulator of metabolism for cellular homeostasis (Zhang, 2015).
The vacuolar (H+)-ATPase (v-ATPase) is an evolutionary-conserved holoenzyme that controls basic cellular processes in eukaryotic cells. As an ATP-dependent proton pump, it acidifies intracellular or extracellular compartments and generates electrochemical gradients, with profound consequences on lysosomal degradation, the transport of metabolites across gut epithelia and many other cellular processes. In lysosomal metabolism, the v-ATPase is a dual player; its proton pumping ability establishes the low pH required by degradative enzymes, whereas its ATPase activity is essential for the aminoacid-dependent activation of TORC1 (the Target Of Rapamycin Complex 1 kinase that links lysosomal degradation to the nutritional state of the cell). Interestingly, in mammalian cell lines, both negative (Settembre, 2011) and positive (Pena-Llopis, 2011) correlation of TORC1 activity with v-ATPase gene expression (ATP6 genes) has been reported. Thus, the effect of TORC1 on the v-ATPase is unclear. However, in both cases, members of the MiT/MITF-family of transcription factors were implicated as mediators of positive or negative regulation by TORC1 (Zhang, 2015).
The four MiT-family genes of mammals, MITF, TFEB, TFE3 and TFEC, encode bHLH-Zip transcription factors that control basic cellular processes in eukaryotes as well as tissue identity and differentiation in animal development. Recent studies in mammalian cell lines have implicated MITF, TFEB and TFE3 in the regulation of degradation pathways. Expression profiling showed induction of lysosomal and autophagy genes by these factors, with most of the targets containing the CLEAR element, a binding sites for TFEB. Interestingly, the nuclear versus cytoplasmic localization of MITF, TFEB, and TFE3 is controlled by the TORC1 kinase through phosphorylation. The mTOR-associated Rag GTPases can interact at an N-terminal motif present in all three MiT factors and promote localization at the lysosome, where phosphorylation of the transcription factors by TORC1 then leads to their cytoplasmic sequestration by the 14-3-3 anchor protein . Alternatively, phosphorylation of TFEB by active TORC1 at a C-terminal serine-rich motif has been proposed to promote its nuclear translocation and activation. Different cell culture conditions and the complication of dealing with multiple MiT family members may have contributed to this discrepancy. Nonetheless, the nature of this TORC1 regulation needs further study (Zhang, 2015).
The invertebrate model organism D. melanogaster offers two advantages. First, it provides a sophisticated genetic model to address questions in vivo, and second, it has a single MiT-family factor. The gene Mitf (CG43369) is expressed broadly at a low level throughout the Drosophila life cycle, but is particularly enriched in the digestive system (Hallsson, 2004). Its physiological roles are unknown, due to a lack of loss-of-function analyses. This study identifies Mitf as a master regulator of the major cellular v-ATPase through transcriptional control of all 15 subunits of the holoenzyme. Modulation of gene expression is direct and impacts holoenzyme activity, with profound consequences on all three metabolic regulators. Mitf, the v-ATPase and TORC1 form a regulatory module that maintains the three factors in dynamic balance and may provide an adaptive feature to its regulation of metabolism. Interestingly, these Mitf functions appear to be conserved in human cells, pointing to an ancient MiT/v-ATPase/TORC1 module for cellular homeostasis (Zhang, 2015).
The role of the v-ATPase as a fundamental regulator of metabolism is well documented
and is underscored by its requirement in all eukaryotic cells (Marshansky, 2014).
Understanding its regulation and how this ties to major metabolic pathways is critical to decoding the complex mechanisms of cellular homeostasis. This study shows that Drosophila Mitf plays a major role in regulating v-ATPase activity. Regulation is at the transcriptional level and direct, through cis sites generally located just upstream of the promoter or in a large early intron of each subunit-encoding Vha locus. Strikingly, fifteen Vha genes appear to be organized into an Mitf-regulated synexpression group that ensures co-production of all components of the major vATPase. Through this mechanism, Mitf functions as a master regulator of the holoenzyme in the
digestive system and other fly tissues (Zhang, 2015).
Concerted expression of v-ATPase loci has also been observed in vertebrates but the genetic and molecular mechanisms mediating this synexpression are largely unknown. Whereas the fly has a single Mitf gene, the situation in mammals is more complex due to the presence of TFEB and TFE3 as well. Nonetheless, in
human melanoma cells and most likely in melanocytes, MITF appears to directly regulate a set of ATP6 genes for all main v-ATPase subunits. Fifteen ATP6 genes (encoding the 13 holoenzyme subunits and 1 accessory protein) are bound in both melanoma cells and primary melanocytes; among these, all show expression correlation with MITF in cell lines and most are downregulated
in response to the partial silencing of MITF in cell culture. These 15 ATP6
genes are considered to be the most likely targets of direct regulation by MITF in melanoma and melanocytes (Zhang, 2015).
The remaining 10 loci show variable effects (with only one bound in melanocytes). These may include genes that are targets of other MiT factors in other tissues and can respond to MITF when it is overexpressed (as is often the case in melanoma tumors). In fact, it is likely that TFEB and TFE3 play a similar role as MITF in controlling most of the ATP6 loci in Hela cells and ARPE-19 cells, respectively. Further analyses of these TFE factors and ATP6 genes in these cell lines will show if this is the case (Zhang, 2015).
In Drosophila and other insects, the v-ATPase works at the plasma membrane of cells
lining gut and Malpighian tubules to regulate pH, energize ion transport and modulate fluid secretion (Wieczorek, 2009). In the developing epithelia of eye, wing and in the ovary, pH modulates the activity of internalized receptors such as Notch; hence, v-ATPase activity has repercussions on signaling. In wing discs, mutations in VhaM8.9 can cause planar cell polarity defects, in addition to disrupting endosomal trafficking (Hermle, 2013). Whereas most of these functions would likely be affected in Mitf mutants,some v-ATPase subunits also fulfill specialized roles (Hiesinger, 2005). In the latter case, the
influence of Mitf would depend on whether the specific subunit is under Mitf control and, if not, on whether the holoenzyme is the critical agent. Ultimately, many of these functions are essential for life and thus explain the lethality of Mitf mutant alleles (Zhang, 2015).
In mammals, the v-ATPase plays essential roles in a broad range of processes that are
regulated by one or other MiT family member. The v-ATPase is essential for the proper function of melanosomes and many melanosome genes, in addition to ATP6 genes, are under MITF control. The v-ATPase also contributes to bone remodeling in osteoclasts, a cell type that expresses, and depends on MITF, TFEB and TFE3 for normal function. Interestingly, double knock-out of the Tfe3 and Mitf genes leads to
osteopetrosis. The v-ATPase has also been found at the plasma
membrane of cancer cells, from where it promotes alkalization of the cytoplasm and acidification of the tumour micro-environment, and this activity was recently linked to the emergence of distant metastases in melanoma. Further studies will elucidate the exact
relationship between MiT factors and the v-ATpase in these contexts (Zhang, 2015).
Importantly, in both vertebrates and Drosophila, the v-ATPase mediates the activation of TORC1 at the lysosomal membrane in response to amino acids (Zoncu, 2011), thereby downregulating lysosomal metabolism. Hence, the v-ATPase can have a negative influence on the lysosome even though it promotes lysosomal function through acidification. In Drosophila, exogenous Mitf leads to increased TORC1 activity and promotes sequestration of Mitf back to the cytoplasm, whereas decreased v-ATPase gene dosage results in more nuclear Mitf as well as lower TORC1 function. In 501mel cell,
exogenous MITF can also increase TORC1 activity. Although the predominant isoform of
MITF in melanocytes does not have the Rag-binding sites, other isoforms do, as do also TFEB
and TFE3. Hence, the regulatory loop is most likely conserved in mammals and functions in many cell types (Zhang, 2015).
Most importantly, Mitf does not merely execute a pro-lysosomal program when freed from
TORC1-induced sequestration. Through the v-ATPase, Mitf feeds back onto TORC1 to promote
and limit the activity of these important metabolic regulators. The Mitf/v-ATPase/TORC1 regulatory
loop adjusts the activity of all three players offering a mechanism for continuously balancing
metabolic pathways as the nutritional state of the cell fluctuates. In addition, it may confer an
adaptive feature to the module. In this model, the level of v-ATPase, present at the
lysosome, would sensitize or desensitize the nutritional sensing mechanism to changes in aa
levels, thereby priming the system to reset at a new normal through TORC1 reactivation or
inactivation. Such mechanism would impose a limit on upregulation of catabolism under lower nutrient conditions, and an upper limit on active TORC1 and its promotion of anabolic pathways
when nutrients are abundant. Interestingly, cell culture experiments show that
prolonged starvation reactivates TORC1; here, the loop offers a potential
molecular mechanism for this effect. Evolutionarily, the Mitf/v-ATPase/TORC1 regulatory module
may have conferred a selective advantage by fine-tuning the nutrient sensing mechanism to
maintain metabolism within an optimal range in an ever-changing environment; an advantage
particularly important for unicellular organisms or for cell types that require more precise metabolic regulation in multicellular ones (Zhang, 2015).
Drosophila provides an excellent metazoan model to investigate the molecular
mechanisms for co-regulation of v-ATPase subunits as well as Mitf's contribution to the
maintenance of cellular homeostasis. It will be also important to investigate how different members
of the MiT family participate in these processes in different cell types under different physiological conditions and what impact they have on cellular homeostasis in health and disease (Zhang, 2015).
The Target of rapamycin (TOR) signaling pathway consists of a set of biochemical processes, which in response to environmental cues and growth factors regulates organismal and cellular growth. TOR is an evolutionarily conserved serine/threonine protein kinase and functions as a core catalytic component of two distinct multiprotein complexes, TOR complex 1 (TORC1) and TORC2. TORC1 controls cell autonomous growth in response to nutrient availability and growth factors, whereas mTORC2 mediates cell proliferation and cell survival by activating several kinases within the AGC family, including AKT, serum and glucocorticoid-regulated kinase (SGK), and protein kinase C (PKC). Although the signaling networks regulated by TORC2 are not completely understood, studies in mice, Drosophila, and Caenorhabditis elegans have demonstrated a role for TORC2 in cell growth. Thus, TORC2 is emerging as a pivotal player in many cancers. As a downstream target of TORC2, AKT plays essential roles in several important cellular processes, including growth, proliferation, survival, and metabolism. Studies in multiple systems have demonstrated that TORC2 inactivates FOXO (forkhead transcription factors of the O-class) through AKT signaling, but it is clear that AKT and FOXO do not mediate TORC2-regulated cell growth. It is therefore critical to identify signaling pathways that act downstream of TORC2 to regulate cell growth (Kuo, 2015).
The myc family of proto-oncogenes encode the transcription factors C-myc, N-myc, and L-myc. Myc proteins play pivotal roles in cell growth and proliferation through the transcriptional regulation of a large number of target genes. As such, dysregulation of Myc contributes to the genesis of many human cancers. Drosophila has a single myc gene (dmyc), which plays a key role in controlling cell size and growth rates by regulating the transcription of mRNAs, ribosomal RNA, and small noncoding RNAs. Hypomorphic dmyc mutations reduce the rate of growth and final size of animals, and dMyc overexpression results in cells and animals that are larger than normal (Kuo, 2015).
Myc has been shown to interact with the TOR pathway. In Drosophila, dMyc is an important mediator of TOR-dependent growth and metabolism, and inhibition of TOR leads to the post-transcriptional down-regulation of dMyc. In mammalian cells, a large-scale quantitative phosphoproteomics study has shown that TOR phosphorylates C-Myc at Ser77. Moreover, similar hypogrowth phenotypes are seen in dmyc, lst8, and rictor mutant animals (the latter two encode essential components of TORC2), suggesting that Myc is an essential link between TORC2 and cell growth (Kuo, 2015).
Using the Drosophila model system, this study found that both cellular and organismal growth defects of dmyc mutant animals were not exacerbated by the loss of LST8. Ectopic expression of dMyc completely rescued the growth defects of both lst8 and rictor mutant animals, including reduced body weight and shrunken eyes and wings. Moreover, the nuclear localization of dMyc was disrupted in lst8 or rictor mutant cells. Furthermore, gene expression profiling revealed that a large set of growth related genes was dysregulated in dmyc, lst8, and rictor mutant animals. These findings suggested that Myc functions downstream of TORC2 to regulate cell growth (Kuo, 2015).
TOR kinase is a highly conserved protein kinase and a central regulator of cell growth. TORC1 has been extensively characterized, but the recent identification of a second TOR complex (TORC2) has complicated the TOR-regulated cell growth pathways. TORC2 regulates growth and metabolism in both mammals and invertebrates. Recent findings indicate that ribosomes physically interact with TORC2 and that this interaction is required for mTORC2 activation. This suggests a critical role for TORC2 in cell growth regulation. However, little is known about the regulation of mTORC2 signaling, or the downstream effectors that implement TORC2-mediated cell growth4. Given evidence that TORC2 regulates AKT/FOXO, AKT/FOXO signaling has been considered the major factor acting downstream of TORC2 to control growth. The current findings that AKT- or FOXO does not affect TORC2-mediated cell growth strongly argue against the AKT/FOXO pathway acting downstream of TORC2 in this context (Kuo, 2015).
The present study found that MYC is required for TORC2-regulated cell growth. Mutations in lst8, rictor, or dmyc had similar growth defects, and lst8 dmyc double mutants did not have more severe growth phenotypes than dmyc mutants. Mosaic analysis in multiple cell types showed that lst8 dmyc double mutant cells had similar growth rates and cell sizes as dmyc mutant cells within the same animals. Moreover, this study established that Myc functions downstream of TORC2 in cell growth. Growth phenotypes associated with loss of TORC2 in the retina, wing, fat body, and the entire body were rescued by the overexpression of dMyc (Kuo, 2015).
Myc protein controls metabolism, cell growth, and proliferation by regulating genes transcribed by RNA Polymerase II, and by stimulating transcription by RNA Polymerases I and III. As Myc is a transcription factor, pathways that regulate the subcellular localization of Myc likely affect its ability to regulate growth and metabolism. This study found that the lack of TORC2 activity is associated the cytoplasmic accumulation of dMyc. Consequently, many Myc target genes were dysregulated in lst8 and rictor mutant tissues. TORC2-mediated nuclear localization of Myc may represent a novel mechanism by which Myc activity is regulated (Kuo, 2015).
As master regulators of cell growth and metabolism, TORC1 and Myc exhibit coordinated patterns of activity. TORC1 activity is required for cancer cell survival, and TORC1 inhibition has remarkable therapeutic efficacy in Myc-driven hematological cancers. In flies, inhibition of TORC1 by molecular inhibitors, genetic manipulations, or starvation leads to the post-transcriptional downregulation of dMyc followed by the repression of dMyc target genes. In mammals, it has been reported that TORC1 activity is required for efficient c-MYC translation in TSC2-null Elt3 rat leiomyoma cells, but opposite results have been reported for colorectal cancer cells, in which TORC1 inhibition by rapamycin treatment or knockdown of Raptor results in phosphorylation and accumulation of Myc. Moreover, in Drosophila intestinal stem cells, excessive TORC1-driven growth in TSC mutants blocked dMyc-induced cell division. These results challenge the notion that TORC1 inhibitors can be used as therapeutic drugs in Myc-driven cancers (Kuo, 2015).
In some cases, such as hyperactivation of AKT signaling, TORC2 is required for proliferation of tumor cells and subsequent tumor growth. The selective requirement for mTORC2 in tumor development suggests that mTORC2 inhibitors may be of substantial clinical utility. The PI3K/AKT signaling cascade is known to regulate metabolic processes via discrete effectors, such as the TSC (tuberous sclerosis) complex and FOXOs. TORC2 inactivates the FOXO branch without affecting the TSC/TORC1 branch. It has been demonstrated that FOXO inhibits MYC function to decrease mitochondrial function and to reduce ROS production. Moreover, a central role for TORC2 in cancer metabolic reprogramming has been proposed, wherein mTORC2 signaling increases cellular c-Myc levels by acetylating FOXO independent of AKT. This study found that TORC2 controlled cell growth by regulating dMyc via nuclear localization. This regulation of dMyc by TORC2 is likely not through TORC2-mediated inactivation of FOXO, because mutations in lst8 or rictor do not affect dMyc transcription or translation. Moreover, previous reports that TORC2 does not regulate cell growth via AKT/FOXO support the model that TORC2 regulates cell growth via Myc independent of FOXO. This finding suggests that TORC2 inhibitors may represent an effective way of treating Myc-driven cancers (Kuo, 2015).
The kinase TOR is found in two complexes, TORC1, involved in growth control, and TORC2 with less well defined roles. This study asked whether TORC2, disrupted by use of Rictor mutant flies, has a role in sustaining cellular stress. TORC2 inhibition in Drosophila was shown to lead to a reduced tolerance to heat stress. Accordingly, upon heat stress, both in the animal and Drosophila cultured S2 cells, TORC2 is activated and is required for the stability of its known target Akt/PKB. The phosphorylation of the stress activated protein kinases is not modulated by TORC2, nor is the heat-induced upregulation of heat shock proteins. Instead, it was shown, both in vivo and in cultured cells, that TORC2 is required for the assembly of heat-induced cytoprotective ribonucleoprotein particles, the pro-survival stress granules. These granules are formed in response to protein translation inhibition imposed by heat stress that appears less efficient in the absence of TORC2 function. It is proposed that TORC2 mediates heat resistance in Drosophila by promoting the cell autonomous formation of stress granules (Jevtov, 2015).
TOR (Target of rapamycin) is a conserved serine/threonine kinase of the PI3K-related kinase family, and functions in two distinct complexes, TOR complex 1 (TORC1) and TOR complex 2 (TORC2). Each complex comprises the kinase along with specific regulatory subunits that give the kinase its functional specificity and structural distinction. The core adaptor proteins of TORC1 are Raptor and LST8, whereas next to LST8, Rictor and Sin1 are the conserved components of TORC2. Removing either of the proteins from a cell destabilizes the TORC2 complex and inhibits its kinase activity. Since its original discovery in screens for rapamycin suppressors, TOR has been extensively studied in the context of TORC1, and has been shown to stimulate key anabolic cellular processes and inhibit the degradative pathway of autophagy with crucial roles in metabolic diseases, cancer and aging . TORC1 is widely regarded as the central node in cell growth control; its activity is dependent on growth factors and nutrient availability, and it is generally shut down in times of stress (Jevtov, 2015).
Unlike TORC1, TORC2 is less well understood and knowledge on upstream cues regulating its activity is scarce. Its role in growth under normal conditions is minor. In lower eukaryotes, TORC2 is activated upon nitrogen starvation, osmotic, heat and oxidative stress and DNA damage, and the TORC2 response to these environmental stresses is related to its likely ancient role in cellular signalling. TORC2 also has a role in actin cytoskeleton rearrangement. Recently, it has also been implicated in gluconeogenesis and sphingolipid metabolism, as well as apoptosis. Protein kinase B (PKB), also known as Akt, a membrane-associated kinase from the family of AGC kinases, with well described roles in cell growth, metabolism and stress, is one of the best characterized downstream targets of TORC2. In vitro, TORC2 has been shown to directly phosphorylate the hydrophobic loop of Akt (S473 in mammals or S505 in Drosophila), thereby increasing its kinase activity (Jevtov, 2015).
There are three well-studied stress response mechanisms in cells. The first is mediated via the stress-activated protein kinases (SAPKs), p38, JNK and Erk, either to protect the cell or to prime it for apoptosis. The second response is the rapid upregulation of transcription of genes encoding heat shock proteins (HSPs) that act as chaperones for cellular proteins to protect them against misfolding and aggregation in stressful conditions. The third includes branches that regulate translation and mRNA turnover. It is well established that heat exposure, oxidative stress and starvation induce the attenuation of bulk protein translation, polysome disassembly and accumulation of untranslated mRNAs. These are stored in cytoplasmic RNA-protein particles (RNP) known as stress granules along with translation initiation factors and RNA-binding proteins. From stress granules, stalled mRNAs can also be transported to the P-bodies (a different type of RNPs that contain RNA decay machinery) for degradation, or upon stress relief, transferred back to polysomes for translation re-initiation. Besides serving as transient protective storage of translation initiation components, the stress granules have also been suggested to serve as a transient station of the SAPKs and other pro-apoptotic kinases under stress, which is regarded to be a protective cellular mechanism against apoptosis. Whether TORC2 acts on these pathways in stress is not known (Jevtov, 2015).
This study shows that TORC2 is specifically required for heat resistance in vivo as Drosophila melanogaster mutants for TORC2 components are selectively sensitive to heat stress. This sensitivity is accompanied by the reduced phosphorylation of Akt due to the loss of the protein itself. Conversely, Akt phosphorylation is enhanced by heat in wild-type Drosophila larvae and cultured cells, showing that TORC2 is activated. Whereas the stress kinase and the HSP branches of the stress response are not affected, we show that the heat-induced stress granule formation is significantly delayed upon loss of TORC2 function, both in cells and in the animals and an reduction of translation inhibition imposed by heat stress might be a cause for this delay. Taken together, it is proposed that under heat stress conditions, TORC2 promotes survival by enabling stress granule assembly (Jevtov, 2015).
The results show that one key branch of the response to heat stress, the formation of stress granules, is delayed by the loss of TORC2 function both in Drosophila tissues and cultured cells. TORC2 is activated upon heat stress and mediates the formation of stress granules, likely required for heat resistance at the cellular level in Drosophila (Jevtov, 2015).
How TORC2 mediates stress granule formation is not clear. Heat stress is known to stimulate the inhibitory phosphorylation of the initiation factor eIF2 αresulting in protein translation stalling. However, this phosphorylation is not required for stress granule formation in Drosophila upon heat stress, so it is unlikely that TORC2 modulates this event (Jevtov, 2015).
This study shows that stress granule formation is delayed by loss of TORC2 function and it is suggested that this is due to a lift on the overall translation inhibition imposed by heat stress, but also under basal conditions. Depletion of TORC2 components appears to stimulate protein translation. This is in accordance with the observations that depletion of either Rictor or Sin1 from Drosophila S2 cultured cells causes their increased proliferation (115%) and cell diameter, respectively. This activation of translation upon loss of TORC2 function could be due to activation of TORC1, as observed previously in Kc cells, another Drosophila cultured cell line. There, depletion of Rictor elevates levels of the phosphorylated 4E-BP, a known target of TORC (Jevtov, 2015).
However, Rictor and Sin1 mutant flies are smaller in size than control animals, suggesting that this translation activation potentially leading to an increase in cell growth and proliferation might be the tissue specific. This might mirror the tissue-specific response in stress granule formation that we report here (Jevtov, 2015).
Such stimulated translation, even upon heat stress might delay or impair stress granule assembly. However, Sin1 depletion has a much stronger effect on translation than Rictor depletion, yet stress granule assembly is inhibited to the same extent in both backgrounds. So whether this lift in translation inhibition is the sole parameter impairing stress granule formation remains to be further investigated. In this regard, Rictor is found at the ribosomes interacting with RACK1, a selective mediator in stress granule function. Thus, it remains to be determined whether TORC2 senses ribosomal activity and mediates the stress granule assembly on its own, rather than indirectly, by providing balance to TORC1 (Jevtov, 2015).
Interestingly, the ribosomal localization of Rictor activates Akt, the TORC2 downstream kinase and this study shows that Akt is activated upon heat stress both in animals and cell lines, in line with mammalian studies. This heat-mediated activation is in line with the finding that S. pombe mutants for Tor1 (kinase of TORC2), Sin1 and Gad8 (Akt ortholog) are also sensitive to heat stress. This suggests that TORC2 - Akt signalling axis represents an ancient and conserved cellular mechanism to cope with heat stress (Jevtov, 2015).
Surprisingly, however, this study found that TORC2 function not only modulates Akt phosphorylation but also its stability. Strikingly, the absence of TORC2 function both in cells and larvae rapidly and significantly obliterates Akt, probably through increased degradation. It is likely not due to a lower translation during stress since translation is less inhibited in heat stress in the absence of TORC2 components. This correlates well with studies in mammalian cells, where PKCalpha, a second known downstream target of TORC2, and a small fraction of Akt are degraded by the proteasome ubiquitin pathway in cells depleted for TORC2 components. This is due to the lack of phosphorylation by TORC2 that primes PKCalpha and Akt for ubiquitination. Whether and how Akt plays a role in stress granule formation in Drosophila remains to be investigated (Jevtov, 2015).
The TORC2 based mechanism that this study proposes is different from the one described in mammalian cells (especially cancer cell lines) where TORC1 is a key player. Indeed, depletion of TORC1 components impairs stress granule assembly by reducing the phosphorylation of 4E-BP, subsequently preventing the formation of eIF4E-eIF4GI cap-dependent translational initiation complexes. In S2 cells, however, this study did not observe a direct role for TORC1 in stress granule formation. Neither Raptor depletion nor rapamycin treatment impairs stress granule formation upon heat stress. Whether this differential involvement of TORC1 and TORC2 in stress granule formation is cell- and tissue- dependent and acts via different pathways remains to be tested. Alternatively, the different mechanisms may suggest that mammals have evolved more sophisticated mechanisms to cope with stress. TORC1 has also been shown to be sequestered in stress granules during heat and other stresses where it suppresses its own apoptotic activity, corroborating its role in stress granule function (Jevtov, 2015).
The importance of studying environmental effects on signalling pathways, like the TOR pathway, is illustrated by the central role of these pathways for progression of diseases, such as metabolic and neurological diseases or cancer. Elucidating the modulation of such pathways under different environmental conditions can potentially identify new targets and processes playing roles in the physiological or pathological regulation of cell survival (Jevtov, 2015).
TORC1 (see Drosophila Tor) is a master regulator of metabolism in eukaryotes that responds to multiple upstream signaling pathways. The GATOR complex is a newly defined upstream regulator of TORC1 that contains two sub-complexes, GATOR1, which inhibits TORC1 activity in response to amino acid starvation and GATOR2, which opposes the activity of GATOR1. The genome of Drosophila contains a single Sea2/Wdr24 homolog encoded by the gene CG7609 that shares 25% identity and 44% similarity to yeast Sea2 and 37% identity and 54% similarity to the human homolog WDR24. This study defines the in vivo role of the GATOR2 component Wdr24 in Drosophila. Wdr24 was shown to have both TORC1 dependent and independent functions in the regulation of cellular metabolism. Through the characterization of a null allele, it was shown that Wdr24 is a critical effector of the GATOR2 complex that promotes the robust activation of TORC1 and cellular growth in a broad array of Drosophila tissues. Additionally, epistasis analysis between wdr24 and genes that encode components of the GATOR1 complex revealed that Wdr24 has a second critical function, the TORC1 independent regulation of lysosome dynamics and autophagic flux. Notably, it was found that two additional members of the GATOR2 complex, Mio and Seh1, also have a TORC1 independent role in the regulation of lysosome function. Wdr24 was also shown to promotes lysosome acidification and autophagic flux in mammalian cells. Taken together these data support the model that Wdr24 is a key effector of the GATOR2 complex, required for both TORC1 activation and the TORC1 independent regulation of lysosomes (Cai, 2016).
In metazoans multiple conserved signaling pathways control the integration of metabolic and developmental processes. TORC1 is an evolutionarily conserved multi-protein complex that regulates metabolism and cell growth in response to an array of upstream inputs including nutrient availability, growth factors and intracellular energy levels. The catalytic component of TORC1 is the serine/threonine kinase Target of Rapamycin (TOR). When nutrients are abundant, TORC1 activity promotes translation, ribosome biogenesis as well as other pathways associated with anabolic metabolism and cell growth. However, when nutrients or other upstream activators are limiting, TORC1 activity is inhibited triggering catabolic metabolism and autophagy (Cai, 2016).
The Seh1 associated/GTPase-activating protein toward Rags (SEA/GATOR) complex is a newly identified upstream regulator of TORC1 that can be divided into two putative sub-complexes GATOR1 and GATOR2 (Bar-Peled, 2013; Dokudovskaya, 2011; Panchaud, 2013). The GATOR1 complex, known as the Iml1 complex or the Seh1 Associated Complex Inhibits TORC1 (SEACIT) in yeast, inhibits TORC1 activity in response to amino acid limitation. SEACIT/GATOR1 contains three proteins Npr2/Nprl2, Npr3/Nprl3 and Iml1/DEPDC5. Recent evidence, from yeast and mammals, indicates that the components of the SEACIT/GATOR1 complex function through the Rag GTPases to inhibit TORC1 activity. Notably, Nprl2 and DEPDC5 are tumor suppressor genes while mutations in DEPDC5 are a leading cause of hereditary focal epilepsies (Cai, 2016).
The GATOR2 complex, which is referred to as Sevh1 Associated Complex Activates TORC1 (SEACAT) in yeast, activates TORC1 by opposing the activity of GATOR1. The SEACAT/GATOR2 complex is comprised of five proteins, Seh1, Sec13, Sea4/Mio, Sea2/WDR24, and Sea3/WDR59. Computational analysis indicates that multiple components of the GATOR2 complex have structural features characteristic of coatomer proteins and membrane tethering complexes. In line with the structural similarity to proteins that influence membrane dynamics, in Drosophila the GATOR2 subunits Mio and Seh1 localize to multiple endomembrane compartments including lysosomes, the site of TORC1 regulation, and autolysosomes. In metazoans, members of the Sestrin and Castor family of proteins bind to and inhibit the GATOR2 complex in response to leucine and arginine starvation respectively. This interaction is proposed to inhibit TORC1 activity through the derepression of the GATOR1 complex. However, how GATOR2 opposes GATOR1 activity, thus allowing for the robust activation of TORC1, remains unknown. Additionally, the role of the GATOR2 complex in the regulation of both the development and physiology of multicellular animals remains poorly defined (Cai, 2016).
Recent evidence from Drosophila indicates that the requirement for the GATOR2 complex may be context specific in multicellular animals. In Drosophila, null alleles of the GATOR2 components mio and seh1 are viable but female sterile. Surprisingly, somatic tissues from mio and seh1 mutants exhibit little if any reductions in cell size and have nearly normal levels of TORC1 activity. In contrast, TORC1 activity is dramatically decreased in ovaries from mio and seh1 mutant females. This decrease in TORC1 activity is accompanied by the activation of catabolic metabolism in the female germ line, a dramatic reduction in egg chamber growth and difficulties maintaining the meiotic cycle. Thus, there is a surprising tissue specific requirement for the GATOR2 components Mio and Seh1 during oogenesis. However, the in vivo role of the other members of the GATOR2 complex in the regulation of cellular metabolism remains undefined (Cai, 2016).
This study defines the in vivo requirement for the GATOR2 component Wdr24 in Drosophila. Wdr24 was found to have two distinct functions. First, Wdr24 is a critical effector of the GATOR2 complex that promotes TORC1 activity and cellular growth in a broad array of tissues. Second, Wdr24 is required for the TORC1 independent regulation of lysosome function and autophagic flux. Notably, two additional members of the GATOR2 complex, Mio and Seh1, also have a TORC1 independent role in the regulation of lysosome function. Taken together these data support the model that multiple components of the GATOR2 complex have both TORC1 dependent and independent roles in the regulation of cellular metabolism (Cai, 2016).
This study describes a dual role for the GATOR2 component Wdr24 in the regulation of TORC1 activity and lysosome dynamics. Wdr24 is a critical effector of the GATOR2 complex that promotes TORC1 activity in both germline and somatic tissues. This lies in contrast to the GATOR2 components Mio and Seh1, which have a limited role in the regulation of TORC1 activity in many cell types. Surprisingly, a second function of Wdr24 was identified that is independent of TORC1 status, the regulation of lysosome acidification and autophagic flux. Taken together these data support the model that the GATOR2 complex regulates both the response to amino acid starvation and lysosome function (Cai, 2016).
Whole animal studies often reveal tissue-specific and/or metabolic requirements for genes that are not readily observed in cell culture. In mammalian and Drosophila tissue culture cells, RNAi based depletions of the GATOR2 components Mio, Seh1, Wdr59, and Wdr24 result in decreased TORC1 activity in return to growth assays (Bar-Peled, 2013, Wei, 2014). These data have resulted in the model that all components of the GATOR2 complex are generally required for TORC1 activation (Wei, 2014). However, the characterization of mio and seh1 null mutants in Drosophila, demonstrated that Mio and Seh1 are critical for the activation of TORC1 and inhibition of autophagy in the female germ line, but play a relatively small role in the regulation of TORC1 activity and autophagy in somatic tissues under standard culture conditions. Thus, the requirement for at least a subset of GATOR2 complex components is tissue and/or context specific (Cai, 2016).
This study reports that the GATOR2 component Wdr24 is required for the full activation of TORC1 in both germline and somatic cells of Drosophila. Consistent with the global down-regulation of TORC1 activity in the absence of Wdr24, wdr24 mutant adults are notably smaller than controls and are female sterile. Depleting the GATOR1 components nprl2 and nprl3 in the wdr24 mutant background rescued the low TORC1 activity, growth defects, and female sterility of wdr24 mutants. Thus, the GATOR2 component Wdr24 is required to oppose GATOR1 activity in both germline and somatic cells of Drosophila. From these results it is proposed that Wdr24 is a key effector of the GATOR2 complex required for the full activation of TORC1 in most cell types (Cai, 2016).
There are several potential models to explain the differential requirement for individual GATOR2 proteins in Drosophila. First, there may be tissue specific requirements for individual GATOR2 subunits. In this model the different phenotypes observed in the seh1 and mio versus wdr24mutants reflects a qualitative difference in the requirement for these proteins in different tissues. However, an alternative model is favored in which Wdr24 is the core effector of GATOR2 activity, with Mio and Seh1 functioning primarily as positive regulators of GATOR2 activity. In this second model, the differential phenotypes observed in the seh1 and mio versus wdr24 mutants reflects a quantitative difference in the requirement for GATOR2 activity in different tissues. The distinction between these two models awaits the identification of the molecular mechanism of Wdr24 and GATOR2 action (Cai, 2016).
A novel TORC1 independent role has been identified for Wdr24 in the regulation of lysosome dynamics and function. In wdr24 mutants, the down-regulation of TORC1 activity and the accumulation of autolysosomes occur independent of nutrient status. It was initially hypothesized that in the absence of the GATOR2 component Wdr24, the deregulation of the GATOR1 complex results in low TORC1 activity, triggering the constitutive activation of autophagy and the accumulation of autolysosomes. Surprisingly, however, epistasis analysis determined that the accumulation of lysosomes could be decoupled from both the chronic inhibition of TORC1 activity and the activation of autophagy. Raising TORC1 activity in the wrd24 mutant background, by depleting either components of the GATOR1 or TSC complex, failed to rescue the accumulation of abnormal lysosomal structures. Notably, it was determined that two additional members of the GATOR2 complex, Mio and Seh1, also regulate lysosomal behavior independent of both GATOR1 and the down-regulation of TORC1 activity. From these data it is inferred that multiple components of the GATOR2 complex have a TORC1 independent role in the regulation of lysosomes (Cai, 2016).
An increased number of autolysosomes is often associated with reduced autophagic flux due to diminished lysosomal degradation. Consistent with reduced autophagic flux, in Drosophila wrd24-/- mutants accumulated enlarged autolysosomes filled with undegraded material. Moreover, lysosomes in the wrd24-/- mutants failed to quench the GFP fluorescence of a GFP-mCherry-Atg8a protein. These phenotypes are consistent with decreased lysosomal pH and degradative capacity. In order to examine in detail the role of Wdr24 in the regulation of lysosome function a wrd24-/- knockout HeLa cell line was generated that recapitulated the phenotypes observed in Drosophila wrd24-/- mutants. Specifically, wrd24-/- HeLa cells had have decreased TORC1 activity and accumulate a large number of autolysosomes. Using multiple assays it was determined that wrd24-/- lysosomes had reduced degradative capacity and autophagic flux and thus accumulate proteins that are normally degraded by lysosomal enzymes such as p62, LC3II and Cathepsin D. Additionally, it was determined that wrd24-/-lysosomes have increased pH relative to wild-type cells, again consistent with reduced lysosomal function. Taken together these data confirm that Wdr24 plays a key role in the regulation of lysosomal activity (Cai, 2016).
This study shows that components of the GATOR2 complex function in the regulation of TORC1 activity and in the TORC1 independent regulation of lysosomal dynamics and autophagic flux. These two functions suggest that the GATOR2 complex may regulate cellular homeostasis by coordinating TORC1 activity with the dynamic regulation of lysosomes during periods of nutrient stress. Intriguingly, several recent reports describe a very similar dual function for the RagA/B GTPases in both mice and zebrafish (Kim, 2014; Shen,2016). RagA/B play a critical role in the activation of TORC1 in the presence of amino acids (Kim, 2008; Sancak, 2008). Surprisingly, however, TORC1 activity was not found to be significantly decreased in cardiomyocytes of RagA/B knockout mice (Cai, 2016).
Nevertheless, the RagA/B mutant cardiomyocytes have decreased autophagic flux and reduced lysosome acidification. From published data, it was concluded that the RagA/B GTPases regulate lysosomal function independent of their role in the regulation of TORC1 activation in some cell types. Similarly, RagA is required for proper lysosome function and phagocytic flux in microglia. Notably, Mio, a component of the GATOR2 complex is found associated with RagA (Bar-Peled, 2013). Thus, in the future it will be important to determine if components of the GATOR2 complex function in a common pathway with the Rag GTPases to regulate lysosomal function (Cai, 2016).
In Saccharomyces cerevisiae single mutants of wrd24/sea2/ and wdr59/sea3 do not exhibit defects in TORC1 regulation but do have defects in vacuolar structure. Moreover, several recently identified genes that regulate the GATOR2-GATOR1-TORC1 pathway in response to amino acid limitation are restricted to metazoans. These data make it tempting to speculate that the ancestral function of the GATOR2 complex maybe the regulation of lysosome/vacuole function and autophagic flux. Indeed, the finding that GATOR2 components regulate lysosome dynamics is particularly intriguing in light of the observation that GATOR2 complex is comprised of proteins with characteristics of coatomer proteins and membrane tethering complexes. Notably, the GATOR2 complex components Mio, Seh1 and Wdr24 localize to lysosomes and autolysosomes. Similarly, these proteins associate with the vacuolar membrane in budding yeast. Thus, going forward it will be important to examine if the GATOR2 complex acts directly on lysosomal membranes to regulate their structure and/or function. More broadly, future studies on the diverse roles of the SEACAT/GATOR2 complex will further understanding of the complex relationship between cellular metabolism and the regulation of endomembrane dynamics in both development and disease (Cai, 2016).
In many metazoans, final adult size depends on the growth rate and the duration of the growth period, two parameters influenced by nutritional cues. In Drosophila, nutrition modifies the timing of development by acting on the prothoracic gland (PG), which secretes the molting hormone ecdysone. When activity of the Target of Rapamycin (TOR), a core component of the nutrient-responsive pathway, is reduced in the PG, the ecdysone peak that marks the end of larval development is abrogated. This extends the duration of growth and increases animal size. Conversely, the developmental delay caused by nutritional restriction is reversed by activating TOR solely in PG cells. Finally, nutrition acts on the PG during a restricted time window near the end of larval development that coincides with the commitment to pupariation. In conclusion, this study shows that the PG uses TOR signaling to couple nutritional input with ecdysone production and developmental timing. Previously studies have shown that the same molecular pathway operates in the fat body (a functional equivalent of vertebrate liver and white fat) to control growth rate, another key parameter in the determination of adult size. Therefore, the TOR pathway takes a central position in transducing the nutritional input into physiological regulations that determine final animal size (Layalle, 2008).
Previous experiments showed that insulin/IGF signaling controls basal levels of ecdysone synthesis in the PG. This, in turn, controls the larval growth rate without modifying the duration of larval growth. These data contrast with the present observations on the role of TOR signaling in the PG and indicate that PG cells discriminate between hormone-mediated activation of InR/PI3K signaling and the nutrient-mediated activation of TOR signaling for the control of ecdysone biosynthesis. Can TOR and InR/PI3K signaling pathways function separately in Drosophila tissues? It has been established both in cultured cells and in vivo that a gain of function for InR/PI3K allows for TORC1 activation through inhibition of TSC2 via direct phosphorylation by AKT/PKB. Such crosstalk between the InR and TOR signaling pathways has important functional implications in cancer cells in which inactivation of the PTEN tumor suppressor leads to an important increase in AKT activity. Nevertheless, the physiological significance of the crosstalk between AKT and TSC2 has been challenged by genetic experiments in Drosophila, leading to the notion that, in the context of specific tissues, TOR and insulin/IGF signaling can be part of distinct physiological regulations for the control of animal growth in vivo. Although not observe in standard conditions, strong InR/PI3K activation in the ring gland shortens larval developmental timing under conditions of food limitation. In light of the present data, this suggests that, in low-food conditions, providing high PI3K activity in PG cells allows for full activation of TOR through the AKT/PKB-mediated inhibitory phosphorylation of TSC2, thus modulating developmental timing. Inversely, a severe downregulation of InR/PI3K signaling in the PG extends larval timing by preventing early larval molts. However, it was observed that strong inhibition of the InR pathway compromises the growth of PG cells, therefore interfering with their capacity to produce normal levels of ecdysone for molting. Overall, previous works as well as the present work highlight the importance of studying signaling networks in the specific contexts (tissue, development) in which these pathways normally operate. This also illustrates that only mild manipulations of these intricate pathways are suitable to unravel the regulatory mechanisms that normally occur within the physiological range of their activities. In conclusion, it is proposed that the insulin/IGF system and TOR provide two separate inputs on PG-dependent ecdysone production: the insulin/IGF system controls baseline ecdysone levels during larval life, and TOR acts upon ecdysone peaks in response to PTTH at the end of larval development (Layalle, 2008).
Important literature describes intrinsic mechanisms controlling a growth threshold for pupariation in insects. After a critical size is attained, the hormonal cascade leading to ecdysone production initiates, and larvae are committed to pupal development, even when subjected to complete starvation. Recent findings in Drosophila by using temperature-sensitive mutants for dInR have revealed that reducing the larval growth rate before the critical size is attained postpones the attainment of this threshold, but has no effect on the final size. Conversely, reducing animals' growth rate after the critical size has been attained leads to strong reduction of the final size. This highlights an important period in the determination of final size, called the terminal growth period (TGP, also called interval to cessation of growth), which spans from the attainment of critical size to the cessation of growth. Due to its exponential rate, growth during that period makes an important contribution to the determination of final size. Interestingly, the duration of the TGP is not affected by the general insulin/IGF system, which explains why reduction of the insulin/IGF system during that period leads to short adults. The present data suggest that the duration of the TGP is an important parameter in the determination of final size that is controlled by TOR. By reducing the level of TOR activity specifically in the PG, neither the growth rate or the critical size for commitment to pupariation is affected. Therefore, the time to attainment of the critical size is not changed. The observation of the developmental transitions in P0206 > TSC1/2 larvae (ectopically expressing TSC1/2) indicate that, indeed, the timing of L1/L2 and L2/L3 molts are not modified. By contrast, the L3/pupa transition is severely delayed, indicating that the interval between attainment of critical size and the termination of growth, i.e., the TGP, is increased. Interestingly, activation of TOR in the PG of fasting larvae leads to a sensible (50%) reduction of the developmental delay induced by low nutrients, whereas it has no effect in normally fed animals. This indicates that the regulation of the TGP by TOR plays an important role in the adaptation mechanisms controlling the duration of larval development under conditions of reduced dietary intake. Other mechanisms, such as the delay to attainment of the critical size due to a reduced growth rate, also contribute to timing of larval development, giving a plausible explanation for the fact that PG-specific TOR activation only partially rescues the increase in larval development timing observed under low-nutrient conditions. Despite characterization in different insect systems, the mechanisms determining the critical size remain to be elucidated. The present study shows that inhibition of TOR signaling in the PG does not modify the minimum size for pupariation. This result is in line with previous findings indicating that nutritional conditions do not modify the critical size in Drosophila. Interestingly, animals depleted of PTTH present an important shift in critical size, indicating that PTTH might participate in setting this parameter. Therefore, mechanisms determining the critical size might reside in the generation or the reception of the PTTH signal, upstream of TOR function in the cascade of events leading to ecdysone production (Layalle, 2008).
What is the limiting step that is controlled by the TOR sensor during the process of ecdysone production? Results obtained by genetic analysis in vivo are reminiscent of in vitro work on dissected PG in the M. sexta model. In these previous studies, PTTH-induced ecdysone production in the PG was shown to induce the phosphorylation of ribosomal protein S6 and was inhibited by the drug rapamycin, later identified as the specific inhibitor of TOR kinase. Interestingly, rapamycin treatment blocked PTTH-induced, but not db-cAMP-induced, ecdysone production, indicating that the drug does not act by simply inhibiting general protein translation in PG cells, but, rather, by inhibiting a specific step controlling PTTH-dependent ecdysone production. More recently, many studies mostly carried out on large insects have started unraveling the response to PTTH in the PG, leading to ecdysone synthesis. No bona fide PTTH receptor is identified yet, and the previously identified response to PTTH is a rise in cAMP, leading to a cascade of activation of kinases, including PKA, MAPKs, PKC, and S6-kinase. S6-kinase-dependent S6 phosphorylation is currently being considered as a possible bottle-neck in the activation of ecdysone biosynthesis by PTTH. The present genetic analysis of ecdysone production in the Drosophila PG now introduces the TOR pathway, the main activator of S6-kinase, as a key controller of ecdysone production and therefore provides a plausible explanation for the rise of S6-kinase in PG cells following PTTH induction. The phenotypes obtained after TOR inhibition in the PG are remarkably similar to the phenotype obtained after ablation of the PTTH neurons. Moreover, ths study shows here that PTTH expression is not altered upon starvation, and that TOR inhibition in PTTH cells has no effect on the duration of larval development, suggesting that PTTH production is not modified by a nutritional stress. Taken together, these data suggest a model whereby limited nutrients induce a downregulation of TOR signaling in the PG, abolish the capacity of PG cells to respond to PTTH and produce ecdysone, and lead to an extension of the terminal growth period (Layalle, 2008).
In conclusion, this study illustrates how the TOR pathway can be used in a specific endocrine organ to control a limiting step in the biosynthesis of a hormone in order to couple important physiological regulations with environmental factors such as nutrition (Layalle, 2008).
Animals use the insulin/TOR signaling pathway to mediate their response to fluctuations in nutrient availability. Energy and amino acids are monitored at the single-cell level via the TOR branch of the pathway and systemically via insulin signaling to regulate cellular growth and metabolism. Using a combination of genetics, expression profiling, and chromatin immunoprecipitation, this study examined nutritional control of gene expression and identified the transcription factor Myc as an important mediator of TOR-dependent regulation of ribosome biogenesis. myc was also identified as a direct target of FOXO, and genetic evidence is provided that Myc has a key role in mediating the effects of TOR and FOXO on growth and metabolism. FOXO and TOR also converge to regulate protein synthesis, acting via 4E-BP and Lk6, regulators of the translation factor eIF4E. This study uncovers a network of convergent regulation of protein biosynthesis by the FOXO and TOR branches of the nutrient-sensing pathway (Teleman, 2008).
The global transcriptional analysis reported in this study has revealed a surprising degree of interconnectedness between the two branches of the nutrient-sensing pathway. Insulin, acting through PI3K and Akt, feeds into the FOXO and TORC1 branches of the pathway, whereas energy levels (AMP/ATP) and amino acids act directly on the TORC1 branch. How are these inputs integrated to maintain energy balance? It was previously known that 4E-BP is transcriptionally regulated by FOXO and posttranslationally regulated by TOR. This study has identified the protein kinase Lk6 as a second direct FOXO target. Thus, there appear to be two parallel, independent mechanisms by which the TOR and FOXO branches of the insulin signaling pathway converge to regulate eIF4E activity and hence cellular protein translation. This 'belt and suspenders' approach to translational control might be important to make the system robust (Teleman, 2008).
A key finding of this study is the identification of Myc as a point of convergent regulation by the FOXO and TOR branches of the pathway. myc mRNA levels are controlled by FOXO in a tissue-specific manner. In addition, Myc protein levels are dependent on TORC1. Why use two independent means to control Myc levels? Transcription alone would limit the speed with which the system can respond to changing nutritional conditions. This might be detrimental, particularly as conditions worsen. Regulation of Myc activity by TORC1 permits a rapid response to changes in energy levels or amino acid availability and could serve to fine tune the nutritional response in the cell by controlling translational outputs. This parallels the situation with 4E-BP, albeit with a slightly different logic. Reduced insulin signaling allows FOXO to enter the nucleus and increase 4E-BP expression and at the same time alleviates TORC1-mediated inhibition of the existing pool of 4E-BP. A subsequent increase in energy or amino acid levels would permit rapid reinhibition of 4E-BP and thus allow a flexible response during the time needed for the pool of protein elevated in response to reduced insulin levels to decay (Teleman, 2008).
In yeast, TORC1 is known to regulate ribosome biogenesis through different nuclear RNA polymerases. It has been shown that yeast TORC1 can bind DNA directly at the 35S rDNA promoter and activate Pol I-mediated transcription in a rapamycin-sensitive manner. Moreover, yeast TORC1 is known regulate Pol II-dependent RP gene expression by controlling the nuclear localization of the transcription factor SFP1 and CRF1, a corepressor of the forkhead transcription factor FHL1. In Drosophila, TORC1 has recently been reported to regulate a set of protein-coding genes involved in ribosome assembly. This study has identified Myc as the missing link mediating TORC1-dependent regulation of this set of genes. Indeed, the fact that more than 90% of TORC1-activated genes contain E boxes suggests that Myc might be the main mediator of this transcriptional program. This connection suggests that expression of Myc targets as a whole should be responsive to nutrient conditions. Indeed, this study found that 33% of direct Myc targets -- defined as genes reported to be bound by Myc when assayed by DNA adenine methyltransferase ID (DamID) in Kc cells and to be regulated by myc overexpression in larvae -- are downregulated upon nutrient deprivation. This is a significant enrichment of 4-fold relative to all genes in the genome, despite the comparison being based on correlating data from different tissue types (Teleman, 2008).
It seems reasonable that cellular translation rates need to be dampened if the TOR branch of the pathway senses low amino acid levels. As ribosome biogenesis is energetically expensive, it may be advantageous to link ribosome biogenesis and translational control via TORC1. This dual regulation is well reflected in tissue growth, since this study observed that Myc, the regulator of ribosome biogenesis, is essential for tissue growth driven by the TOR pathway but not sufficient to drive growth in the absence of TOR activity. The FOXO branch of the pathway senses reduced insulin or mitogen levels. FOXO is also highly responsive to oxidative and other stresses and would integrate this information into the cellular control of translation. The data support the notion of a network in which TOR and FOXO regulate protein biosynthesis by converging on Myc to regulate ribosome biogenesis and on eIF4E activity via 4E-BP and Lk6 to regulate translation initiation (Teleman, 2008).
The work presented in this study complements a previous study in which larvae were either starved completely or starved for amino acids only, while having a supply of energy in the form of sugar. A significant and positive correlation (~0.4) indicates general agreement between the two data sets, but they differ in two ways. The current goal was to explore the regulatory network by which insulin controls cellular transcription. Individual tissues were isolated rather than assaying the whole animal. Genes found to be regulated in a previous but not in the current assays may be regulated in tissues other than muscle or adipose tissue. Conversely, genes identified only by the current study might be regulated oppositely in different tissues or might only be regulated in a subset of tissues and so be missed in a whole-animal analysis.
Is Myc also involved in nutritional signaling networks in mammals? No similar rapid downregulation of c-myc was seen in response to rapamycin in human cell lines, suggesting that the mechanism by which TOR signaling controls gene expression may differ between phyla. This is further supported by the fact that the sets of genes reported to be rapamycin regulated also appear to be largely distinct in Drosophila and mammalian cells, with the caveat that different cell types were used in the two analyses. Although the mechanism does not appear to be identical in mammals, there are several suggestions in the literature of a connection between c-Myc and nutritional signaling. For example, dMyc and c-Myc share the ability to regulate ribosome biogenesis, although the specific target genes through which they do so are different. There is also evidence that mammalian c-myc expression in liver is regulated by nutrition and that transgenic expression of c-myc in liver affects metabolism, i.e., glucose uptake and gluconeogenesis. Furthermore, it has been reported that FOXO3 represses Myc activity in colon cancer cells by inducing members of the Mad/Mxi family, which are known to antagonize Myc. The current data suggest that Max and Mnt are not transcriptionally regulated by insulin or FOXO in Drosophila, whereas myc is. This is similar to what has been reported in murine lymphoid cells, in which c-myc expression is regulated by the FOXO homolog FKHRL1. These parallels between the fly and mammalian systems suggest a broader connection between insulin signaling and activity of the Myc/Mnt/Max network. Although some features may be different in the two systems, the similarities merit further investigation (Teleman, 2008).
Finally, this work has revealed a surprising amount of tissue specificity in the transcriptional response to insulin signaling. Roughly half of the genes regulated by insulin in adipose tissue or in muscle were not significantly regulated in the other tissue. Furthermore, 155 genes were differentially regulated in the two tissues (i.e., upregulated in one tissue and downregulated in the other). This likely reflects the roles of the different tissues in the organism's response to nutrient deprivation. Further work will elucidate the underlying molecular mechanisms (Teleman, 2008).
The TOR and Jak/STAT signal pathways are highly conserved from Drosophila to mammals, but it is unclear whether they interact during development. The proline-rich Akt substrate of 40 kDa (PRAS40) mediates the TOR signal pathway through regulation of TORC1 activity, but its functions in TOR complex 1 (TORC1, a rapamycin-sensitive form of Tor in mice that consists of mTOR, raptor, and mLST8) proved in cultured cells are controversial. The Drosophila gene Lobe (L) encodes the PRAS40 ortholog required for eye cell survival. L mutants exhibit apoptosis and eye-reduction phenotypes. It is unknown whether L regulates eye development via regulation of TORC1 activity. This study found that reducing the L level, by hypomorphic L mutation or heterozygosity of the null L mutation, resulted in ectopic expression of unpaired (upd), which is known to act through the Jak/STAT signal pathway to promote proliferation during eye development. Unexpectedly, when L was reduced, decreasing Jak/STAT restored the eye size, whereas increasing Jak/STAT prevented eye formation. Ectopic Jak/STAT signaling and apoptosis are mutually dependent in L mutants, indicating that L reduction makes Jak/STAT signaling harmful to eye development. In addition, genetic data suggest that TORC1 signaling is downregulated upon L reduction, supporting the idea that L regulates eye development through regulation of TORC1 activity. Similar to L reduction, decreasing TORC1 signaling by dTOR overexpression results in ectopic upd expression and apoptosis. A novel finding from these data is that dysregulated TORC1 signaling regulates the expression of upd and the function of the Jak/STAT signal pathway in Drosophila eye development (Wang, 2009).
The target of rapamycin (TOR) and Jak/STAT signal pathways are highly conserved in animals and important in many developmental processes. Dysregulation of these pathways can lead to cancer formation. This study presents data showing that TOR regulates the function of Jak/STAT signaling during Drosophila eye development (Wang, 2009).
The gene unpaired (upd) encodes a ligand that activates Drosophila Jak/STAT signaling. It is expressed in the posterior margin of the dorsal/ventral (D/V) boundary, the posterior center (PC), in the larval eye imaginal disc at second and early third instar stages. Notch at the D/V boundary activates the transcription of eye gone (eyg), which activates upd expression at the PC. Expression of upd is also regulated by Hedgehog (Hh) signaling. The cells of Drosophila compound eyes are derived from the eye-antennal disc, which develops from ectoderm of the embryo and grows inside the larva. These cells proliferate rapidly during the first and second instar stage. In early third instar larvae, morphogenetic furrow (MF) that arise at the posterior margin progresses in a wave-like manner toward the anterior margin of the eye disc. Jak/STAT signaling is known to promote proliferation during eye development, and is required for MF initiation; a loss of Jak/STAT function results in reduced eyes. Therefore, Jak/STAT signaling is regulated by Notch/Eyg and the Hh signaling pathways, and plays positive roles in eye development (Wang, 2009).
TOR signaling is one of the downstream branches of insulin signal pathway. Insulin and insulin-like growth factor elicit a signal cascade involving phosphatidyl-inositol 3-kinase (PI3K) that stimulates PDK-mediated Akt phosphorylation. Phosphorylated Akt can activate TOR, which nucleates the TOR complex 1 (TORC1), allowing it to phosphorylate the downstream targets, the translational repressor eukaryotic initiation factor (4EBP) and the ribosomal protein S6 kinase (S6k). Phosphorylation of 4EBP and S6K promotes CAP-dependent translation and thereby increases protein synthesis. In addition, activation of TOR can also promote ribosome biogenesis via Myc. Loss of the Drosophila TOR (dTOR) function reduces eye size, indicating that TOR signaling is required for eye development (Wang, 2009).
PRAS40 mediates the insulin signal pathway from Akt to TORC1. Upon insulin stimulation, activated Akt phosphorylates PRAS40 and causes it to dissociate from TORC1, allowing TORC1 signaling to proceed. Thus, PRAS40 can apparently act as an inhibitor of TORC1. However, it has been reported that PRAS40 is required for TORC1 activity, and thus the interactions of PRAS40 with TORC1, based on studies in cultured cells are controversial. The effect of PRAS40 on TORC1 signaling in vivo is still unclear (Wang, 2009).
The Drosophila Lobe (L) protein shares high sequence conservation with PRAS40. L mutants have reduced adult eyes and exhibit ectopic apoptosis during eye development, indicating that L is required for eye development. But whether it regulates eye development via regulation of TORC1 activity is unknown (Wang, 2009 and references therein).
This study identified a new L allele, Lfee. Quantitative RT-PCR and genetic analysis revealed that Lfee is a hypomorphic allele. The eye defect was mediated by ectopic Jak/STAT signaling and cell apoptosis. In L mutants, the ectopic Jak/STAT signaling had a negative effect on eye development, but not a positive one as previously reported. It was also found that TORC1 signaling was hypoactivated in L mutants, suggesting that, like PRAS40, L is required for TORC1 activity. This study suggests that hypoactivated TORC1 signaling in L mutants result in ectopic Jak/STAT signaling and apoptosis, impairing eye development (Wang, 2009).
The spontaneous mutant fly, freaky eye (fee), is homozygously viable and has abnormal adult eyes. The eyes of most fee flies are smaller than those of wild type flies because of a nick at the anterior border of the eye. At the nicked region, extra hairs and/or rod-like tissues are usually present. Overgrowth of eye tissue occasionally occurs, resulting in eye enlargement. The eyes of fee flies were categorized into six classes depending on their size relative to the eyes of the wild type. The various eye-reduction phenotypes of fee flies were similar to those of L mutants. For example, the Lsi heterozygote exhibits slightly reduced eyes that are nicked near the anterior D/V boundary, similar to the major fee phenotype. In the Lsi homozygote, the ventral eye is absent, which is also reminiscent of the fee phenotype (Wang, 2009).
Whether fee is a mutant of L was investigated; the trans-heterozygotes for fee and the null mutant Lrev6-3 had smaller eyes than fee flies. In addition, fee could to be recombined with Lrev6-3, suggesting that fee is allelic to L. Quantitative RT-PCR showed that the L mRNA levels were highly reduced in fee flies, suggesting that fee is a L hypomorphic mutant; therefore these were designated Lfee (Wang, 2009).
This study shows that reduction of L phenocopies overexpression of dTOR. Overexpression of dTOR has been reported to produce phenotypes similar to that of loss of dTOR, because excess dTOR may titrate cofactors and thereby decrease TOR activity. This suggests that TOR signaling is downregulated by L reduction. Consistent with this, genetic analysis of L mutants and TOR signal pathway component suggest TORC1 hypoactivity in L mutants. PRAS40 has been proposed to function in the assembly of TORC1. It is possible that, in similar way to dTOR overexpression, reducing L impairs TORC1 assembly, thus decreasing TORC1 signaling. Reduction of L may disrupt eye development through downregulation of TORC1 signaling, supporting the idea that PRAS40 is required for TORC1 activity (Wang, 2009).
Drosophila eye development requires the TOR and Jak/STAT signal pathways, but it is not know whether an interaction between these two signal pathways occurs. Endogenous upd expression is present in the posterior center (PC) of the eye disc, but not in the interior eye disc. This study demonstrated that L reduction can induce ectopic upd expression in the interior eye disc, indicating that L is a negative regulator for upd expression. The data show that L reduction-mediated eye disruption is due to hypoactivation of TORC1 signaling, suggesting that hypoactivity of TORC1 is responsible for inducing upd expression (Wang, 2009).
Ectopic upd expression is induced by reduction of L (Lfee and Lrev/+), but not by its complete loss (Lrev homozygous clones), suggesting that different L levels may cause distinct effects. As PRAS40 acts to transmit the Akt signal to TORC1, complete loss, but not reduction, of L could result in an uncoupling between Akt and TORC1. This would release the Akt-mediated inhibition of TORC1, resulting in increased TORC1 activity. Thus, complete loss of L or PRAS40 may increase TORC1 activity. It is possible that the opposite functions of PRAS40 reported in cultured cells could be due to different PRAS40 levels remaining after knockdown. Whether complete loss of L function inhibits or promotes TORC1 signaling in Drosophila eyes remains to be investigated (Wang, 2009).
Mosaic analysis data showed that dTOR homozygote clones did not induce ectopic upd expression, suggesting that complete loss of dTOR function has a different effect from that of L reduction. Overexpression of dMyc can completely restore the eye size in the Lfee flies, but only partially represses the eye defect of dTOR overexpression. These data support the idea that L reduction may not equate to loss of dTOR. It was reasoned that as TOR is involved in TORC1 and TORC2, its loss should eliminate the functions of both TORC1 and TORC2. Because L participates only in TORC1 signaling, reduction of L would affect TORC1 signaling only. The regulation of TORC1 and TORC2 signaling by L needs further investigation (Wang, 2009).
It was found that suppressing apoptosis can decrease ectopic upd expression upon L reduction, suggesting that apoptosis is a cause of ectopic upd expression. It has been reported that apoptosis can activate ectopic upd expression and Jak/STAT signaling via Notch signaling in apoptosis-induced compensatory proliferation. However, ectopic upd expression on L reduction is not likely to be mediated by Notch activity, and no ectopic proliferation occurs. Thus, apoptosis due to L reduction is different from apoptosis-induced compensatory proliferation. Further, TOR hypoactivation may trigger ectopic upd expression independent of apoptosis; suppression of apoptosis did not eliminate all ectopic upd expression. Further investigation of how hypoactivated TORC1 regulates upd expression is needed (Wang, 2009).
The Drosophila Upd acts through Jak/STAT signaling to promote proliferation during eye development. However, this study found that on L reduction, decreasing Jak/STAT signaling could restore the eye defect, whereas increasing the upd expression level could completely abolish eye development. Thus, an unexpected finding was that ectopic Jak/STAT signaling in L mutants is harmful to eye development (Wang, 2009).
The fact that decreasing Jak/STAT signaling can reduce apoptosis in L mutants indicates that the induction of ectopic Jak/STAT signaling is required for apoptosis. It was reasoned that the apoptosis-promoting ability of Jak/STAT is possibly due to its repression of Serrate (Ser) expression. Ser expression is inhibited by L mutation, and loss of Ser function during eye development causes apoptosis. The current data showed that heterozygosity for Ser can reduce eye size in Lfee heterozygotes, but not in the wild type, suggesting that decreased Ser expression may play a role in eye reduction. Whether Ser repression mediates the apoptosis remains to be investigated. In addition, because inhibition of apoptosis does not strongly restore the L eye defect, but decreasing Jak/STAT activity fully restores it (comparing ey > p35 and Stat92Ets), there is the possibility that the ectopic Jak/STAT activity affects eye development via an apoptosis-independent mechanism. Thus, a novel finding from the data is that Jak/STAT signaling can negatively regulate eye development (Wang, 2009).
An important issue is the control over the positive and negative roles of Jak/STAT signaling during eye development. Overexpression of upd driven by ey-GAL4 in the wild type produces adult with enlarged eyes, but it eliminates eye formation in L mutants. Because L reduction exhibits hypoactivation of TORC1 signaling, it is speculated that TORC1 signaling plays a role in controlling the balance between the opposing functions of Jak/STAT signaling (Wang, 2009).
In summary, reduction of the Drosophila PRAS40 L results in hypoactivation of TORC1 signaling. This leads to apoptosis and ectopic Jak/STAT activation, both of contribute to disruption of eye development. The data indicate that TORC1 signaling is able to regulate the expression and functions of the Jak/STAT signal pathway during eye development. Further studies using L mutants may uncover the mechanisms by which L regulates TORC1 signaling, and how TOR controls the Jak/STAT signal pathways. Also noteworthy is the report that decreasing PRAS40 can increase apoptosis of tumor cells, and it is therefore of interest to investigate whether PRAS40 and TORC1 can regulate the Jak/STAT signal pathway in tumors (Wang, 2009).
To cover the receptive field completely and non-redundantly, neurons of certain functional groups arrange tiling of their dendrites. In Drosophila class IV dendrite arborization (da) neurons, the NDR family kinase Tricornered (Trc) is required for homotypic repulsion of dendrites that facilitates dendritic tiling. This study reports that Sin1, Rictor, and target of rapamycin (TOR), components of the TOR complex 2 (TORC2), are required for dendritic tiling of class IV da neurons. Similar to trc mutants, dendrites of sin1 and rictor mutants show inappropriate overlap of the dendritic fields. TORC2 components physically and genetically interact with Trc, consistent with a shared role in regulating dendritic tiling. Moreover, TORC2 is essential for Trc phosphorylation on a residue that is critical for Trc activity in vivo and in vitro. Remarkably, neuronal expression of a dominant active form of Trc rescues the tiling defects in sin1 and rictor mutants. These findings suggest that TORC2 likely acts together with the Trc signalling pathway to regulate the dendritic tiling of class IV da neurons, and thus uncover the first neuronal function of TORC2 in vivo (Koike-Kumagai, 2009).
Many stem, progenitor and cancer cells undergo periods of mitotic quiescence from which they can be reactivated. The signals triggering entry into and exit from this reversible dormant state are not well understood. In the developing Drosophila central nervous system, multipotent self-renewing progenitors called neuroblasts undergo quiescence in a stereotypical spatiotemporal pattern. Entry into quiescence is regulated by Hox proteins and an internal neuroblast timer. Exit from quiescence (reactivation) is subject to a nutritional checkpoint requiring dietary amino acids. Organ co-cultures also implicate an unidentified signal from an adipose/hepatic-like tissue called the fat body. This study provides in vivo evidence that Slimfast amino-acid sensing and Target of rapamycin (TOR) signalling activate a fat-body-derived signal (FDS) required for neuroblast reactivation. Downstream of this signal, Insulin-like receptor signalling and the Phosphatidylinositol 3-kinase (PI3K)/TOR network are required in neuroblasts for exit from quiescence. Nutritionally regulated glial cells provide the source of Insulin-like peptides (ILPs) relevant for timely neuroblast reactivation but not for overall larval growth. Conversely, ILPs secreted into the haemolymph by median neurosecretory cells systemically control organismal size but do not reactivate neuroblasts. Drosophila thus contains two segregated ILP pools, one regulating proliferation within the central nervous system and the other controlling tissue growth systemically. These findings support a model in which amino acids trigger the cell cycle re-entry of neural progenitors via a fat-body-glia-neuroblasts relay. This mechanism indicates that dietary nutrients and remote organs, as well as local niches, are key regulators of transitions in stem-cell behaviour (Sousa-Nunes, 2011).
In fed larvae, Drosophila neuroblasts exit quiescence from the late first instar (L1) stage onwards. This reactivation involves cell enlargement and entry into S phase, monitored in this study using the thymidine analogue 5-ethynyl-2'-deoxyuridine (EdU). Reactivated neuroblast lineages (neuroblasts and their progeny) reproducibly incorporated EdU in a characteristic spatiotemporal sequence: central brain --> thoracic --> abdominal neuromeres. Mushroom-body neuroblasts and one ventrolateral neuroblast, however, are known not to undergo quiescence and to continue dividing for several days in the absence of dietary amino acids. This indicates that dietary amino acids are more than mere 'fuel', providing a specific signal that reactivates neuroblasts. However, explanted central nervous systems (CNSs) incubated with amino acids do not undergo neuroblast reactivation unless co-cultured with fat bodies from larvae raised on a diet containing amino acids. Therefore the in vivo requirement for a fat-body-derived signal (FDS) in neuroblast reactivation was tested by blocking vesicular trafficking and thus signalling from this organ using a dominant-negative Shibire dynamin (SHIDN). This strongly reduced neuroblast EdU incorporation, indicating that exit from quiescence in vivo requires an FDS. One candidate tested was Ilp6, known to be expressed by the fat body, but neither fat-body-specific overexpression nor RNA interference of this gene significantly affected neuroblast reactivation. Fat-body cells are known to sense amino acids via the cationic amino-acid transporter Slimfast (SLIF), which activates the TOR signalling pathway, in turn leading to the production of a systemic growth signal. Fat-body-specific overexpression of the TOR activator Ras homologue was shown to be enriched in brain (RHEB), or of an activated form of the p110 PI3K catalytic subunit, or of the p60 adaptor subunit, had no significant effect on neuroblast reactivation in fed animals or in larvae raised on a nutrient-restricted diet lacking amino acids. In contrast, global inactivation of Tor, fat-body-specific Slif knockdown or fat-body-specific expression of the TOR inhibitors Tuberous sclerosis complex 1 and 2 (Tsc1/2) all strongly reduced neuroblasts from exiting quiescence. Together, these results show that a SLIF/TOR-dependent FDS is required for neuroblasts to exit quiescence and that this may be equivalent to the FDS known to regulate larval growth (Sousa-Nunes, 2011).
Next, the signalling pathways essential within neuroblasts for their reactivation were investigated. Nutrient-dependent growth is regulated in many species by the interconnected TOR and PI3K pathways. In fed larvae, it was found that neuroblast inactivation of TOR signalling (by overexpression of TSC1/2), or PI3K signalling (by overexpression of p60, the Phosphatase and tensin homologue PTEN, the Forkhead box subgroup O transcription factor FOXO or dominant-negative p110), all inhibited reactivation. Conversely, stimulation of neuroblast TOR signalling (by overexpression of RHEB) or PI3K signalling [by overexpression of activated p110 or Phosphoinositide-dependent kinase 1 (PDK1)] triggered precocious exit from quiescence. RHEB overexpression had a particularly early effect, preventing some neuroblasts from undergoing quiescence even in newly hatched larvae. Hence, TOR/PI3K signalling in neuroblasts is required to trigger their timely exit from quiescence. Importantly, neuroblast overexpression of RHEB or activated p110 in nutrient-restricted larvae, which lack FDS activity, was sufficient to bypass the block to neuroblast reactivation. Notably, both genetic manipulations were even sufficient to reactivate neuroblasts in explanted CNSs, cultured without fat body or any other tissue. Together with the previous results this indicates that neuroblast TOR/PI3K signalling lies downstream of the amino-acid-dependent FDS during exit from quiescence (Sousa-Nunes, 2011).
To identify the mechanism bridging the FDS with neuroblast TOR/PI3K signalling, the role of the Insulin-like receptor (InR) in neuroblasts was tested. Importantly, a dominant-negative InR inhibited neuroblast reactivation, whereas an activated form stimulated premature exit from quiescence. Furthermore, InR activation was sufficient to bypass the nutrient restriction block to neuroblast reactivation. This indicates that at least one of the potential InR ligands, the seven ILPs, may be the neuroblast reactivating signal(s). By testing various combinations of targeted Ilp null alleles and genomic Ilp deficiencies, it was found that neuroblast reactivation was moderately delayed in larvae deficient for both Ilp2 and Ilp3 (Df(3L)Ilp2-3) or lacking Ilp6 activity. Stronger delays, as severe as those observed in InR31 mutants, were observed in larvae simultaneously lacking the activities of Ilp2, 3 and 5 [Df(3L)Ilp2-3, Ilp5] or Ilp1-5 [Df(3L)Ilp1-5]. Despite the developmental delay in Df(3L)Ilp1-5 homozygotes, neuroblast reactivation eventually begins in the normal spatial pattern -- albeit heterochronically -- in larvae with L3 morphology. Together, the genetic analysis shows that Ilp2, 3, 5 and 6 regulate the timing but not the spatial pattern of neuroblast exit from quiescence. However, as removal of some ILPs can induce compensatory regulation of others, the relative importance of each cannot be assessed from loss-of-function studies alone (Sousa-Nunes, 2011).
Brain median neurosecretory cells (mNSCs) are an important source of ILPs, secreted into the haemolymph in an FDS-dependent manner to regulate larval growth. They express Ilp1, 2, 3 and 5, although not all during the same development stages. However, this study found that none of the seven ILPs could reactivate neuroblasts during nutrient restriction when overexpressed in mNSCs. Similarly, increasing mNSC secretion using the NaChBac sodium channel or altering mNSC size using PI3K inhibitors/activators, which in turn alters body growth, did not significantly affect neuroblast reactivation under fed conditions. Surprisingly, therefore, mNSCs are not the relevant ILP source for neuroblast reactivation. Nonetheless, Ilp3 and Ilp6 messenger RNAs were detected in the CNS cortex, at the early L2 stage, in a domain distinct from the Ilp2+ mNSCs. Two different Ilp3-lacZ transgenes indicate that Ilp3 is expressed in some glia (Repo+ cells) and neurons (Elav+ cells). An Ilp6-GAL4 insertion indicates that Ilp6 is also expressed in glia, including the cortex glia surrounding neuroblasts and the glia of the blood-brain barrier (BBB) (Sousa-Nunes, 2011).
Next the ability of each of the seven ILPs to reactivate neuroblasts when overexpressed in glia or in neurons was assessed. Pan-glial or pan-neuronal overexpression of ILP4, 5 or 6 led to precocious reactivation under fed conditions. Each of these manipulations also bypassed the nutrient restriction block to neuroblast reactivation, as did overexpression of ILP2 in glia or in neurons, or ILP3 in neurons. In all of these ILP overexpressions, and even when ILP6 was expressed in the posterior Ultrabithorax domain, the temporal rather than the spatial pattern of reactivation was affected. Importantly, experiments blocking cell signalling with SHIDN indicate that glia rather than neurons are critical for neuroblast reactivation. Interestingly, glial-specific overexpression of ILP3-6 did not significantly alter larval mass. Thus, in contrast to mNSC-derived ILPs, glial-derived ILPs promote CNS growth without affecting body growth (Sousa-Nunes, 2011).
Focusing on ILP6, CNS explant cultures were used to demonstrate directly that glial overexpression was sufficient to substitute for the FDS during neuroblast exit from quiescence. In vivo, ILP6 was sufficient to induce reactivation during nutrient restriction when overexpressed via its own promoter or specifically in cortex glia but not in the subperineurial BBB glia, nor in many other CNS cells that were tested. Hence, cortex glia possess the appropriate processing machinery and/or location to deliver reactivating ILP6 to neuroblasts. Ilp6 mRNA is known to be upregulated rather than downregulated in the larval fat body during starvation and, accordingly, Ilp6-GAL4 activity is increased in this tissue after nutrient restriction. Conversely, it was found that Ilp6-GAL4 is strongly downregulated in CNS glia during nutrient restriction. Thus, dietary nutrients stimulate glia to express Ilp6 at the transcriptional level. Consistent with this, an important transducer of nutrient signals, the TOR/PI3K network, is necessary and sufficient in glia (but not in neurons) for neuroblast reactivation. Together, the genetic and expression analyses indicate that nutritionally regulated glia relay the FDS to quiescent neuroblasts via ILPs (Sousa-Nunes, 2011).
This study used an integrative physiology approach to identify the relay mechanism regulating a nutritional checkpoint in neural progenitors. A central feature of the fat-body --> glia --> neuroblasts relay model is that glial insulin signalling bridges the amino-acid/TOR-dependent fat-body-derived signal (FDS) with InR/PI3K/TOR signalling in neuroblasts. The importance of glial ILP signalling during neuroblast reactivation is also underscored by an independent study, published while this work was under revision (Chell, 2010). As TOR signalling is also required in neuroblasts and glia, direct amino-acid sensing by these cell types may also impinge upon the linear tissue relay. This would then constitute a feed-forward persistence detector, ensuring that neuroblasts exit quiescence only if high amino-acid levels are sustained rather than transient. This study also showed that the CNS 'compartment' in which glial ILPs promote growth is functionally isolated, perhaps by the BBB, from the systemic compartment where mNSC ILPs regulate the growth of other tissues. The existence of two functionally separate ILP pools may explain why bovine insulin cannot reactivate neuroblasts in CNS organ culture, despite being able to activate Drosophila InR in vitro. Given that insulin/PI3K/TOR signalling components are highly conserved between insects and vertebrates, it will be important to address whether mammalian adipose or hepatic tissues signal to glia and whether or not this involves an insulin/IGF relay to CNS progenitors. In this regard, it is intriguing that brain-specific overexpression of IGF1 can stimulate cell-cycle re-entry of mammalian cortical neural progenitors, indicating utilization of at least part of the mechanism identified by this study in Drosophila (Sousa-Nunes, 2011).
B cell lymphoma 2 (Bcl-2) proteins are the central regulators of apoptosis. The two bcl-2 genes in Drosophila modulate the response to stress-induced cell death, but not developmental cell death. Because null mutants are viable, Drosophila provides an optimum model system to investigate alternate functions of Bcl-2 proteins. This report explores the role of one bcl-2 gene in nutrient stress responses. Starvation of Drosophila larvae lacking the bcl-2 gene buffy decreases survival rate by more than twofold relative to wild-type larvae. The buffy null mutant reacted to starvation with the expected responses such as inhibition of target of rapamycin (Tor) signaling, autophagy initiation and mobilization of stored lipids. However, the autophagic response to starvation initiated faster in larvae lacking buffy and was inhibited by ectopic buffy. Unusually high basal Tor signaling, indicated by more phosphorylated S6K, was detected in the buffy mutant, and removal of a genomic copy of S6K, but not inactivation of Tor by rapamycin, reverted the precocious autophagy phenotype. Instead, Tor inactivation also required loss of a positive nutrient signal to trigger autophagy and loss of both was sufficient to activate autophagy in the buffy mutant even in the presence of enforced phosphoinositide 3-kinase (PI3K) signaling. Prior to starvation, the fed buffy mutant stored less lipid and glycogen, had high lactate levels and maintained a reduced pool of cellular ATP. These observations, together with the inability of buffy mutant larvae to adapt to nutrient restriction, indicate altered energy metabolism in the absence of buffy. All animals in their natural habitats are faced with periods of reduced nutrient availability. This study demonstrates that buffy is required for adaptation to both starvation and nutrient restriction. Thus, Buffy is a Bcl-2 protein that plays an important non-apoptotic role to promote survival of the whole organism in a stressful situation (Monserrate, 2012).
This study has demonstrated that buffy is required for normal larval responses to nutrient stress. This could not be attributed to a role for buffy in sensing nutrient starvation and activating normal starvation responses. Instead, larvae lacking buffy displayed characteristics of altered energy metabolism and increased growth signaling through Tor, as demonstrated by increased phosphorylated S6K. This study did not address whether the increased Tor signaling is a cause or result of the energy metabolism of the buffy mutant. It is conceivable that upregulation of Tor signaling results in increased energy consumption to promote growth. However, it was not observed that the increased phosphorylated S6K was correlated with increased growth in the buffy mutant, suggesting that the Tor signaling was balanced by the altered energy metabolism in the mutant. Taking into account the current understanding of Bcl-2 proteins, it is postulated that Buffy is required to maintain energy homeostasis at a set point that is optimal for both growth and starvation responses. Loss of buffy results in a change of this homeostatic set point that may directly or indirectly upregulate growth signaling and that places the animal closer to a metabolic cliff in terms of its ability to survive nutrient stress (Monserrate, 2012).
In investigating starvation responses in the buffy mutant, it was observed that fat body autophagy was initiated faster in buffy mutant larvae. Although Tor signaling normally inhibits autophagy, the high level of phosphorylated S6K maintained by the mutant was required for precocious starvation-induced autophagy. Since autophagy is a mechanism to recycle essential building blocks when nutrients in the environment are scarce, whether reduced energy storage was correlated with precocious autophagy was investigated. Wild-type animals, with normal Tor signaling, provided with 20% of the normal nutrients (20% CY, 1.8% sucrose) were autophagic after 2 h of starvation. But this nutrient-restriction diet resulted in a much greater reduction in stored nutrients in the fat body than the 15% reduction in lipid storage observed in the buffy mutant. In addition, excess growth signaling by ectopic activation of Tor signaling in wild-type larvae, was not sufficient to induce precocious autophagy. It is proposed that the unique combination of an altered metabolism and increased Tor signaling in larvae lacking buffy that render the animal more sensitive to nutrient stress and results in precocious autophagy (Monserrate, 2012).
Energy sensing has been linked to autophagy initiation in mammals. ULK1 (mammalian ATG1) function is regulated by both Tor and AMPK. In the simplest current thinking, nutrient deprivation both inactivates Tor and activates AMPK to phosphorylate and activate ULK1 to initiate autophagy. In Drosophila, the complex of ATG1/ATG13 is regulated by Tor and AMPK is required for starvation-induced autophagy, suggesting that regulation of autophagy initiation by phosphorylation is similar in fruit flies. In larvae lacking buffy, decreased cellular energy might more efficiently activate ATG1/ATG13, possibly mediated through AMPK. This model does not take into account that precocious autophagy in the buffy mutant required phosphorylated S6K. There is conflicting data as to the role of S6K in autophagy. Because inhibition of Tor induces autophagy, phosphorylation of S6K is inversely correlated with autophagy. However, S6K has been shown to be required for starvation-induced autophagy in Drosophila, and plays a positive role in autophagic induction in mammals. Faster autophagy in the buffy mutant may reflect a positive signaling role for S6K in autophagy initiation that contributes to this phenotype. Indeed it is intriguing to postulate that a metabolic signal from loss of the positive nutrient signal is transmitted through phosphorylated S6K in all animals, and that augmented phosphorylated S6K merely potentiates this signal in the buffy mutant (Monserrate, 2012).
The metabolism phenotypes observed in the buffy mutant larvae (smaller energy stores in the fat body, increased glucose utilization inferred from less glycogen storage, a reduced pool of ATP and increased lactate) are most simply explained by a shift in the balance of glycolysis to oxidative phosphorylation toward glycolysis. Glycolysis is less efficient at generating ATP and increased glycolysis generates excessive pyruvate that is converted to lactate. To maintain glycolysis at a higher rate, a higher percentage of ingested glucose and lipids must be shuttled into glycolysis at the expense of storage in the fat body. Animals that rely more on glycolysis for energy generation would certainly be more sensitive to nutrient restriction. This hypothesis is supported by recent evidence that oxygen consumption and cellular ATP levels were reduced, while glycolysis was increased, in Bcl-2-associated X protein (BAX)-deficient cells. Two recent studies on Bcl-xL also support direct regulation of oxidative phosphorylation: one demonstrated that Bcl-xL controls the levels of the metabolite acetyl coenzyme A (acetyl-CoA) and the other proposed that neuronal Bcl-xL directly regulates the efficiency of ATP synthesis by the F1F0 ATP synthase complex. Consistent with less efficient oxidative phosphorylation, buffy mutant larvae are sensitive to the reactive oxygen species (ROS) generator, paraquat, and have a twofold increase in ROS. Increased ROS has also been reported to result from enforced Tor signaling in Drosophila . Intriguingly, ROS has been proposed to affect S6K phosphorylation
(Monserrate, 2012).
Bcl-2 proteins govern permeabilization of the mitochondrial outer membrane that leads to loss of mitochondrial energy production and release of apoptogenic factors such as cytochrome c. Buried within the vast quantity of publications investigating Bcl-2 proteins are studies that support a role for some of the Bcl-2 proteins in mitochondrial energetics, generally with a focus on ectopic expression of Bcl-2 proteins and effects on metabolism with regard to apoptosis. Many studies have shown an interaction between Bcl-2 proteins and the voltage-dependent anion channel (VDAC) that regulates movement of metabolites between the mitochondria and the cytosol. Although this interaction is not required for mitochondrial-dependent cell death, it may be that Bcl-2 proteins modulate mitochondrial energetics through VDAC. One of the metabolites whose uptake is facilitated by VDAC is Ca2+. Intracellular Ca2+ signaling is regulated by the ER and Bcl-2 proteins influence ER calcium content through modulation of the inositol triphosphate receptor (IP3R) and the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA). Uptake of Ca2+ released by the ER can stimulate mitochondrial energy metabolism through several targets. Ectopic Buffy decorates both the mitochondria and the ER in various cell types, leaving open the possibility that Buffy has a functional role in ER-mitochondria Ca2+ signaling. Additionally, Bcl-2 proteins play a role in mitochondrial morphogenesis, both in the fragmentation observed upon apoptosis induction. Mitochondria in Drosophila also fragment prior to cell death. This study observed that buffy mutant fat body had a higher density of mitochondria that were in general smaller and less 'snake like'. However, buffy mutant animals did not have more mitochondria since no increase in mitochondrial genomes was observed in larval or fat body extracts (Monserrate, 2012).
This study has demonstrated that Drosophila larvae lacking buffy are sensitive to nutrient restriction and starvation. buffy mutant larvae have unusual basal characteristics: increased Tor signaling, reduced energy source storage, reduced ATP levels and increased lactate levels. The data provides evidence that, in the normal animal, Buffy maintains basal energy homeostasis to enable appropriate responses to nutrient stress. Future studies will determine how Buffy influences basal energy metabolism and clarify the relationship between energy metabolism and S6K regulation. The recent reports demonstrating that Bcl-xL regulates metabolic efficiency in neurons and that Bax promotes bioenergetics in HCT-116 cells and primary hepatocytes support the hypothesis that some Bcl-2 proteins have a non-apoptotic role to produce resistance to stressors by maintaining mitochondrial energetics. The current data adds to these reports, and is unique because it investigates the effect on organismal health of loss of a bcl-2 gene and provides evidence for crosstalk with Tor signaling. It is important to note that the Drosophila Bcl-2 proteins are bona fide Bcl-2 proteins containing BH1-4 domains and a C-terminal transmembrane domain, have the ability to bind other Bcl-2 proteins and can substitute for their mammalian counterparts (Monserrate, 2012).
Apoptosis is most often considered at the cellular level: cells that are unnecessary, damaged or diseased are removed by cell suicide. But it is essential to keep in mind that apoptosis promotes survival of the entire organism. It is certainly plausible that the same proteins that function as a rheostat for apoptosis also perform a similar function for survival, through energy modulation, in stressful life situations that are normally encountered by the organism (Monserrate, 2012).
Amino-acid starvation leads to an inhibition of cellular proliferation and the induction of programmed cell death (PCD) in the Drosophila ovary. Disruption of insulin signaling has been shown to inhibit the progression of oogenesis, but it is unclear whether this phenotype mimics starvation. This study investigated whether the insulin-mediated phosphoinositide kinase-3 pathway regulates PCD in mid oogenesis. It was reasoned that under well-fed conditions, disruption of positive components of the insulin signaling pathway within the germline would mimic starvation and produce degenerating egg chambers. Surprisingly, mutants did not mimic starvation but instead produced many abnormal egg chambers in which the somatic follicle cells disappeared and the germline persisted. These abnormal egg chambers did not show an induction of caspases and lysosomes like that observed in wild-type (WT) degenerating egg chambers. Egg chambers from insulin signaling mutants were resistant to starvation-induced PCD, indicating that a complete block in insulin-signaling prevents the proper response to starvation. However, target of rapamycin (Tor) mutants did show a phenotype that mimicked WT starvation-induced PCD, indicating an insulin independent regulation of PCD via Tor signaling. These results suggest that inhibition of the insulin signaling pathway is not sufficient to regulate starvation-induced PCD in mid oogenesis. Furthermore, starvation-induced PCD is regulated by Tor signaling converging with the canonical insulin signaling pathway (Pritchett, 2012).
These results indicate that the insulin signaling pathway is an important factor for cell survival in the Drosophila ovary, similar to its role in mammals. Mammalian ovaries cultured in serum-free media show an induction in apoptotic and autophagic PCD. Survival of mouse primordial follicles requires PI3K, phosphoinositide-dependent protein kinase-1 (PDK1), and S6k1, which are members of the insulin signaling cascade. Treatment with rapamycin, a drug known to block Tor activity, inhibits oocyte growth in cultured Drosophila and mammalian ovaries, and leads to apoptosis and autophagy. Taken together, these findings suggest an evolutionarily conserved role for insulin and Tor signaling in promoting survival in the ovary. Furthermore, characterization of starvation-induced PCD in the Drosophila ovary may give insight into the mechanisms of degeneration of defective oocytes in mammalian systems during reproductive aging and fertility disorders (Pritchett, 2012).
Organs often need to coordinate the growth of distinct tissues during their development. This study analyzed the coordination between germline cysts and the surrounding follicular epithelium during Drosophila oogenesis. Genetic manipulations of the growth rate of both germline and somatic cells influence the growth of the other tissue accordingly. Growth coordination is therefore ensured by a precise, two-way, intrinsic communication. This coordination tends to maintain constant epithelial cell shape, ensuring tissue homeostasis. Moreover, this intrinsic growth coordination mechanism also provides cell differentiation synchronization. Among growth regulators, PI3-kinase and TORC1 also influence differentiation timing cell-autonomously. However, these two pathways are not regulated by the growth of the adjacent tissue, indicating that their function reflects an extrinsic and systemic influence. Altogether, these results reveal an integrated and particularly robust mechanism ensuring the spatial and temporal coordination of tissue size, cell size, and cell differentiation for the proper development of two adjacent tissues (Vachias, 2014: PubMed).
Several main conclusions can be drawn from this work. First, in each follicle, growth is intrinsically coordinated between the two tissues. Second, this growth control tends to optimize cell shape in the epithelium. This is likely to be representative of the development of many epithelia where cell shape must be maintained because it is essential for the function of the tissue. In the third place, growth control has a very important impact on differentiation timing in each tissue. Furthermore, several growth pathways can cell-autonomously influence differentiation rate but are not regulated by the adjacent tissue, indicating that they only respond to extrinsic cues. Finally, as a whole, this study reveals the robustness of the spatiotemporal pattern allowing the production of mature eggs with a normal shape and a normal size. At least two examples based on Pten somatic clones can illustrate this robustness. WT border cells migrate perfectly 'on time' in a follicle in which mutant somatic cells have induced a faster germline development. Second, a WT looking mature egg can be found in the middle of an ovariole, suggesting that all developmental steps have been faster but correctly orchestrated. This robustness probably reflects the fact that final egg size is constant, that most of the developmental steps have to occur at a specific size, and that differentiation is able to block growth when the definitive egg size is reached. These observations raise the question as to how the differentiation program regulates growth and especially growth arrest in each follicle (Vachias, 2014).
These results indicate a two-way communication between the germline and the soma to ensure their coordination. It was also observed that somatic cells can influence other somatic cells but, importantly, that such an effect depends on the relay of the germ cells. This result suggests that coordination is achieved by different signals depending on the tissue. The soma and germline could communicate via the secretion of growth factors controlling the adjacent tissue, though obvious candidates were excluded. An alternative explanation would be that, as it is proposed in mammals, the two tissues are interdependent for specific metabolites, although it would be independent of TORC1, a classical sensor of metabolic activity. Finally, an attractive hypothesis would be that growth regulation between the soma and the germline depends on a mechanical steady state. Germline growth creates a tension on the follicle cell, leading to the proposal that this tension could trigger epithelial growth. If so, it would also mean that follicle cells provide a mechanical strain limiting germline growth. The mechanical control of growth in epithelial cells is usually devoted to the Hippo pathway, which is not involved in this instance. Thus, this work does not allow favoring one or the other of these nonexclusive mechanisms (Vachias, 2014).
Altogether, these results highlight several dimensions of coordination between cell growth, cell shape, and cell identity and all this between two distinct tissues. These different functional links offer a highly robust program in space and time. The relevance for such robustness has been very recently highlighted because it probably confers the reproducibility on embryonic development. Since usual pathways controlling growth are not involved in this two-way communication, this multidimensional coordination will be a useful framework for identifying molecular actors ensuring tissue homeostasis in the recurrent context of the development of two adjacent tissues (Vachias, 2014).
How adipocytes contribute to the physiological control of stem cells is a critical question towards understanding the link between obesity and multiple diseases, including cancers. Previous studies have revealed that adult stem cells are influenced by whole-body physiology through multiple diet-dependent factors. For example, nutrient-dependent pathways acting within the Drosophila ovary control the number and proliferation of germline stem cells (GSCs). The potential role of nutrient sensing by adipocytes in modulating stem cells in other organs, however, remains largely unexplored. This study report that amino acid sensing by adult adipocytes specifically modulates the maintenance of GSCs through a Target of Rapamycin-independent mechanism. Instead, reduced amino acid levels and the consequent increase in uncoupled tRNAs trigger activation of the GCN2-dependent amino acid response pathway within adipocytes, causing increased rates of GSC loss. These studies reveal a new step in adipocyte-stem cell crosstalk (Armstrong, 2014).
The TOR protein kinases (TOR1 and TOR2 in yeast; mTOR/FRAP/RAFT1 in mammals) promote cellular proliferation in response to nutrients and growth factors, but their role in development is poorly understood. The Drosophila TOR homolog dTOR is required cell autonomously for normal growth and proliferation during larval development, and for increases in cellular growth caused by activation of the phosphoinositide 3-kinase (PI3K) signaling pathway. As in mammalian cells, the kinase activity of dTOR is required for growth factor-dependent phosphorylation of S6 kinase in vitro, and overexpression of S6k in vivo can rescue dTOR mutant animals to viability. Loss of dTOR also results in cellular phenotypes characteristic of amino acid deprivation, including reduced nucleolar size, lipid vesicle aggregation in the larval fat body, and a cell type-specific pattern of cell cycle arrest that can be bypassed by overexpression of the S-phase regulator cyclin E. These results suggest that dTOR regulates growth during animal development by coupling growth factor signaling to nutrient availability (Zhang, 2000).
The Drosophila genome encodes four members of the PIK-related family: mei41, which encodes an ATR/ATM homolog required for cell cycle arrest in response to DNA damage (Hari, 1995); CG6535, a second ATM-related gene of unknown function; CG4549, whose closest relative encodes the nonsense-mediated mRNA decay protein SMG-1 in C. elegans, and the dTOR. A fifth member of this family, the DNA-dependent protein kinase, is not found in Drosophila or C. elegans (Zhang, 2000).
To begin a mutational analysis of dTOR, the Berkeley Drosophila Genome Project P-element database was searched and two independent homozygous lethal lines bearing P insertions in the dTOR gene were identified (designated here as dTORP1 and dTORP2). Mobilization of these elements restores viability to each chromosome, indicating that the insertions are responsible for the associated lethality. Comparison of the insertion sites of dTORP1 and dTORP2 with the dTOR transcription unit reveal that the P-elements are inserted at 24 and 74 bases downstream of the dTOR transcription start site, respectively, and would likely interfere with normal dTOR expression (Zhang, 2000).
To generate additional dTOR alleles, a series of deletions spanning the dTOR gene was generated by imprecise mobilization of the P-elements. One such mutant, designated dTORDeltaP, was selected for further analysis. Sequence analysis reveals that the dTORDeltaP deletion originates at the dTORP2 insertion site and extends 3514 bp downstream, removing the dTOR translation start site and amino-terminal 902 codons, and thus likely represents a null allele of dTOR. This was confirmed by the absence of detectable dTOR protein in immunoblots of dTORDeltaP larval extracts. A 9.4-kb genomic rescue construct encompassing the dTOR gene and no other predicted transcription units restores full viability and fertility to dTORDeltaP homozygotes (Zhang, 2000).
dTORDeltaP homozygotes hatch at normal rates, but grow more slowly than normal and eventually arrest during larval development, reaching only 24% the mass of wild-type controls. Larvae homozygous for dTORP1 or dTORP2 alleles display a less severe phenotype, eventually growing to approximately 40% and 79% the mass of wild type, respectively, indicating that these alleles likely retain partial dTOR function. In each case, the mutants remain viable and active during an extended larval period of ~30 d, and eventually die without pupating. Larvae heterozygous for dTOR grow at a rate indistinguishable from wild-type controls under normal culture conditions, but are hypersensitive to low concentrations of rapamycin. Thus, dTOR encodes a rapamycin-sensitive protein required for normal growth during larval development (Zhang, 2000).
Overall growth of an organism is generally accompanied by increases in cell number (proliferation), cell size (hypertrophy), or both. To determine how mutations in dTOR inhibit growth, these parameters were examined in a number of tissues. The effect of dTOR on cell size was analyzed in marked clones of dTORDeltaP homozygous cells, which were generated by FLP/FRT-mediated mitotic recombination in dTORDeltaP heterozygous animals. Examination of adult cuticular structures reveal that dTOR homozygous mutant cells are markedly reduced in size. For example, bristles of the wing margin that lack dTOR were both thinner and shorter than adjacent wild-type cell. Area measurements of mutant clones in the wing epithelium show that dTORDeltaP mutant cells are approximately half (56%) the size of controls. Similar effects have been observed in the eye, abdomen, and notum (Zhang, 2000).
To determine whether loss of dTOR affects the size of actively proliferating cells, dTOR mutant clones were examined in the developing imaginal discs, epithelial primordia that proliferate mitotically to give rise to adult structures. Imaginal wing discs containing GFP-marked clones of dTORDeltaP homozygous cells were dissociated into single cells, which were then analyzed by flow cytometry. The mean forward light scatter value (a measure of cell size) of dTOR mutant cells is decreased by 30% compared to wild-type control cells from the same discs. This decrease in cell size is observed in all phases of the cell cycle. Thus, loss of dTOR causes a cell autonomous reduction in the size of both proliferating and postmitotic cells (Zhang, 2000).
Fluorescence-activated cell sorter (FACS) analysis has also revealed that the cell cycle phasing of dTOR cells differs significantly from that of controls, with relatively more cells in G1, and fewer in S and G2 phases. This is consistent with the ability of rapamycin to induce G1 arrest in yeast and in mammalian cell culture. To measure proliferation rates of dTOR mutant cells, the number of cells in dTORDeltaP clones were compared with that of their wild-type sister clones (twin spots). Clones of dTORDeltaP mutant cells are similar in size to their twin spots at 48 h after induction, but by 72-96 h they contain significantly fewer cells, indicating that loss of dTOR leads to a reduced rate of cell proliferation. In addition, lone twin spots lacking a corresponding mutant sister clone have occasionally been observed at 96 h after induction, indicating that at some frequency dTORDeltaP homozygous cells are eliminated from the disc epithelium. Because dTORDeltaP cells remain viable for weeks in the context of a homozygous mutant animal, the loss of dTORDeltaP mutant clones is likely the result of cell competition with adjacent wild-type cells, which is known to occur for cells with a growth disadvantage (Zhang, 2000).
Growth properties of cells in the salivary glands of homozygous dTORDeltaP larvae were also examined. The salivary gland is comprised of two cell types: polytene gland cells that undergo multiple rounds of endoreduplication to generate giant nuclei with a ploidy of up to 2048 C, and imaginal ring cells that remain diploid and cycle mitotically. Loss of dTOR affects both cell types. The endoreplicative cells in dTORDeltaP salivary glands undergo only four to five rounds of replication before entering quiescence, reaching a ploidy of 16-32C and a size ~10% that of wild type. The imaginal rings in dTORDeltaP larvae contain approximately fivefold fewer cells than wild type. Together, these results indicate that dTOR is required to promote cell cycle progression in both mitotic and endoreplicative cells, and acts primarily at the G1/S transition (Zhang, 2000).
The cell autonomous reduction in the size of dTOR mutant cells is reminiscent of mutations in components of the PI3K/S6K signaling pathway. Mutations in Pten, the fly homolog of the PTEN tumor suppressor, cause activation of this pathway, leading to increased cell growth. To determine whether dTOR is required for PI3K-dependent signaling, the growth properties of cells lacking both Pten and dTOR were examined. Clonal loss of Pten causes enlargement of imaginal and adult cells, and increases the percentage of cells in the S and G2 phases of the cell cycle. In contrast, cells carrying null alleles of both Pten and dTOR are indistinguishable from cells lacking dTOR alone, with a similar reduction in cell size and accumulation in G1. Loss of dTOR also prevents the increased proliferation caused by mutations in Pten. Cells mutant for weaker alleles of Pten and dTOR (MGH1 and P2, respectively) are intermediate in size, indicating that dTORP2 cells retain partial signaling function. It is concluded that dTOR is epistatic to Pten, and therefore, that dTOR functions at a step downstream of or in parallel to PI3K signaling (Zhang, 2000).
In further tests of dTOR's role in PI3K/S6k signaling, it was found that rapamycin inhibits the serum-dependent phosphorylation of Drosophila p70S6k (S6k) expressed in S2 cells. Dephosphorylation of S6k by rapamycin is prevented by cotransfection of a dTOR point mutant containing a Ser1956 to Thr substitution (dTORRR, which confers rapamycin resistance to mammalian and yeast TOR proteins. Expression of dTORRR carrying an additional point mutation in a residue crucial for kinase activity (dTORRRKD fails to protect S6k from rapamycin-induced dephosphorylation, indicating that the kinase function of dTOR is required to maintain S6k phosphorylation (Zhang, 2000).
To determine whether these biochemical interactions between dTOR and S6k are relevant to their functions in vivo, tests were performed for genetic interactions between them. Remarkably, constitutive overexpression of Drosophila S6k or human p70S6K1 is able to rescue dTORP2/P2 and dTORP1/P2 flies to viability. The greatest degree of rescue is provided by a mutant version of p70S6K1, in which four mitogen-induced phosphorylation sites are mutated to aspartate and glutamate residues (mutant D4). Expression of this construct allowed 74% of expected dTORP1/P2 progeny to survive to adulthood, whereas no dTORP1/P2 animals survived in the absence of S6k overexpression. dTOR flies rescued by S6k overexpression are slightly smaller than wild-type controls, but are fertile and develop at a similar rate as wild type. Although overexpression of S6k does not rescue dTORDeltaP null mutants to adulthood, it does enable them to progress to the pupal stage. Overexpression of S6k in wild-type larvae also confers significant resistance to rapamycin. Again, constitutively active p70S6K1 provides the greatest degree of rapamycin resistance. Together, these results indicate that a major function of dTOR is to maintain levels of active S6k sufficient for normal growth (Zhang, 2000).
Like dTOR mutants, wild-type larvae deprived of amino acids enter an extended larval period with little or no growth. Amino acid deprivation also causes a series of distinctive cellular phenotypes including a reduction in nucleolar area, changes in morphology of the larval fat body, and a cell type-specific cell cycle arrest. Since TOR proteins have been proposed to be regulated in response to amino acid levels, it was of interest to examine whether loss of dTOR mimics these cellular effects (Zhang, 2000).
The nucleolus is the major cellular site of ribosomal assembly, and its size has been shown to correlate with protein synthetic capacity and proliferation rate. To measure nucleolar size in wild-type and dTOR mutant cells, wing imaginal discs containing clones of dTORDeltaP homozygous cells were labeled with an antibody against the nucleolar protein fibrillarin, and examined by confocal sectioning. The nucleolar area in clones of dTOR mutant cells in the wing imaginal disc is approximately half that of surrounding wild-type cells, consistent with a role for dTOR in ribosome biogenesis (Zhang, 2000).
During metamorphosis or starvation, stores of protein, lipid, and glycogen are mobilized from adipose cells of the larval fat body and are used by other tissues as an energy source in place of dietary nutrients. These metabolic effects are visible as changes in appearance of fat body cells. The major visible change in fat body cells in larvae deprived of amino acids is an aggregation of lipid vesicles, and this effect is indistinguishable from that caused by loss of dTOR (Zhang, 2000).
Within 48-72 h of amino acid withdrawal, endoreplicative cells become quiescent, whereas mitotic neuroblasts of the central nervous system continue to cycle for at least 8 d in the absence of amino acids. This pattern of cell cycle responses is distinct from that caused by complete inhibition of protein synthesis, which causes all larval cells to arrest DNA synthesis. To determine whether loss of dTOR causes a cell cycle response similar to that elicited by starvation, cell cycle behavior of these cell types was examined in dTORDeltaP larvae at multiple stages of development. At 3-4 d after egg deposition (AED), both endoreplicative and mitotic tissues were found to cycle normally in dTORDeltaP homozygotes, as measured by incorporation of the nucleotide analog BrdU. In contrast, by 5-6 d AED all endoreplicative tissues including the gut, fat body, and salivary glands fail to incorporate BrdU, whereas neuroblasts continued to cycle. A similar pattern is observed at 10 d AED. Presumably the appearance of this cell cycle arrest at 4-5 d AED results from the perdurance of maternal stores of dTOR mRNA or protein until this time. The cell cycle arrest of dTOR endoreplicative cells can be bypassed by overexpression of the G1/S regulators cyclin E or dE2F/dDP, as has been demonstrated previously for endoreplicative cells arrested by amino acid withdrawal. Thus, amino acid insufficency and loss of dTOR each cause similar growth arrests, changes in cell morphology, and cell type-specific patterns of G1 arrest (Zhang, 2000).
In budding yeast, TOR proteins govern S-phase entry in response to nutrient levels by regulating translation of the G1/S regulator Cln3 (Barbet, 1996 and Polymenis, 1997). Drosophila cyclin E has been proposed to play a role analogous to Cln3, and its abundance increases in response to growth stimuli such as overexpression of activated Ras. Because cyclin E overexpression is able to bypass the cell cycle arrest in dTOR mutants, whether loss of dTOR affects cyclin E expression was examined. Immunoblot analysis of whole larval extracts has revealed that the level of cyclin E protein is reduced ~30-fold in dTORDeltaP mutants, as compared to wild-type larvae of similar stage (Zhang, 2000).
Clonal induction of cells lacking cyclin E in the wing imaginal disc results in a G1 arrest within one to two cell divisions. In contrast, cells mutant for dTOR continue to cycle slowly for several days, and give rise to clones containing multiple cells, indicating that dTOR mutant cells retain at least partial cyclin E activity. Accordingly, cyclin E protein is reduced but not eliminated in dTOR mutant clones in the wing disc. Although 72-h dTORDeltaP clones containing little or no detectable cyclin E immunoreactivity are often observed, many dTOR clones were found containing cells with apparently normal cyclin E levels. It is concluded that the observed reduction in cyclin E protein levels in dTORDeltaP larval extracts is due largely to reduced cyclin E levels in endoreplicating cells, which comprise the majority of larval mass, resulting in a G1 arrest in this cell type. In contrast, dTOR may be required in imaginal cells to maintain normal rates of cyclin E accumulation, rather than absolute levels, consistent with the reduced rate of cell division and extended G1 phase observed in dTOR mutant clones (Zhang, 2000).
dTOR mutants differ from the known PI3K pathway mutants in several important respects. (1) The growth defects caused by loss of dTOR are more severe than those arising from mutations in components of the PI3K signaling pathway. (2) Null mutations in the PI3K subunits Dp110 or p60 allow growth to the third instar larval stage, and chico (IRS-1) and S6k mutants survive to adulthood. At least in the case of chico and S6k, many cells are able to cycle normally throughout development. In contrast, animals lacking dTOR reach only the size of second instar larvae, at which point they undergo cell cycle arrest. (3) Whereas overexpression of Dp110, Akt, or S6k leads to increased growth rate and cell size, dTOR overexpression inhibits growth and reduces cell size. Although variations in genetic background may partially account for some of these phenotypic differences, these results are inconsistent with dTOR acting as an integral component of a linear PI3K/Akt/S6k pathway, and instead argue that dTOR may converge upon this pathway in response to a distinct set of cues (Zhang, 2000 and references therein).
In yeast, TOR1 and TOR2 regulate cell growth directly in response to levels of nutrients such as amino acids, rather than in response to intercellular signals. Similarly, human mTOR may also be regulated by nutrient levels. These considerations prompted a comparison of the phenotypes of dTOR mutant animals with the physiological changes caused by nutrient deprivation. By the three criteria examined -- nucleolar size, fat body vesicle formation, and endoreplicative cell cycle arrest -- loss of dTOR precisely mirrors the effects of starvation. An efficient explanation of these results is that dTOR is required for normal responses to changes in nutrient levels. This would be consistent with a model in which full activation of growth targets such as S6k requires two distinct inputs: growth factor-mediated intercellular signals through PI3K, and nutrient-sensing signals through TOR. In this view, TOR proteins may act as part of a checkpoint to attenuate growth factor signaling when local conditions are unfavorable for cell growth (Zhang, 2000).
How TOR proteins might be regulated by nutrient levels is unclear. In the case of amino acids, recent evidence suggests that the primary signal may be uncharged tRNA, which increases in abundance when amino acid levels are low. Interestingly, other members of the PIK-related kinase family such as ATM and DNA-PK also act as checkpoint proteins that are regulated by specific nucleic acid structures. In addition, a recently described member of this family, SMG-1, is involved in the degradation of aberrant mRNAs containing inappropriate nonsense codons. Thus, regulation by nucleic acid may be a common feature of this kinase family (Zhang, 2000 and references therein).
Activation of p70S6K is a common response to virtually all mitogenic stimuli. Phosphorylation of the target of p70S6K, ribosomal protein S6, leads to the selective increase in translation of a subset of transcripts that contain an oligopyrimidine tract at their 5' termini (5' TOP). This class of messages encodes ribosomal proteins and translation elongation factors, and thus p70S6K activation leads to increased ribosomal biogenesis. The demonstration in this study that overexpression of S6k can rescue dTOR mutants to viability indicates that S6k is a critical effector of dTOR, and that one essential function of dTOR is to regulate the activity of S6k (Zhang, 2000).
Despite the central role of S6k in mediating the functions of dTOR, several lines of evidence indicate that dTOR has additional S6k-independent roles. (1) dTOR mutant phenotypes are more severe than those of S6k; lack of dTOR results in growth arrest at the second instar larval stage, whereas S6k mutant animals survive to adulthood, albeit with a delayed development and decreased body size. (2) Although S6k overexpression suppresses the lethality of dTOR mutants, this is only the case for hypomorphic dTOR allelic combinations; dTOR null animals expressing S6k advance only to the pupal stage. (3) dTOR flies rescued by S6k do not grow to the full size of wild-type controls. Thus, some dTOR functions are not fully rescued by S6k. As noted above, in mammalian cells mTOR also stimulates translation through phosphorylation and inactivation of 4E-BP1, an inhibitory binding factor of the translation initiation factor eIF4E. Drosophila eIF4E mutants display a severe growth arrest phenotype, and thus aspects of dTOR function that are not rescued by S6k may reflect diminished eIF4E activity. However, neither overexpression of eIF4E nor mutations in 4E-BP1 detectably alleviate dTOR mutant phenotypes (Zhang, 2000).
In addition to effects on translation, studies in budding yeast have found that inactivation of TOR modulates the level of a number of growth-related or nutrient-regulated transcripts, including those encoding ribosomal proteins (Barbet, 1996; Zaragoza, 1998; Beck, 1999; Cardenas, 1999; Hardwick, 1999; Powers, 1999). A transcriptional role for TOR in higher eukaryotes has been reported as well. Moreover, recent studies have found that mTOR can interact with a number of additional signaling factors including STAT3, protein kinase C, c-Abl, and 14-3-3 (Parekh, 1999; Kumar, 2000a and b; Mori, 2000; Yokogami, 2000). Rapamycin has also recently been shown to disrupt microtubule assembly and function in yeast, independent of its effects on translation (Choi, 2000). Thus, the inability of S6k overexpression to fully supplant dTOR may be due to a requirement for dTOR in multiple cellular functions (Zhang, 2000).
The reduced growth of dTOR mutants reflects reductions in both cell size and cell number. Because direct inhibition of proliferation increases rather than decreases cell size, it is proposed that the primary function of dTOR is to promote cell growth, and that the decreased proliferation of dTOR mutant cells is a secondary effect in response to their reduced rate of growth. The accumulation of dTOR mutant cells in the G1 phase of the cell cycle suggests that the G1/S transition is particularly sensitive to growth rate. This is consistent with previous observations that stimulation of cell growth by overexpression of dMyc, PI3K, or activated Ras promotes progression through the G1/S but not G2/M transition (Zhang, 2000 and references therein).
A factor likely to be involved in coupling growth and division rates is the G1/S regulator cyclin E. Cyclin E is rate limiting for G1/S progression in imaginal discs, and its levels increase by a post-transcriptional mechanism in response to stimulation of cell growth. The demonstration that dTOR is required for normal accumulation of cyclin E suggests that translational control is likely to play an important role in cyclin E regulation. In budding yeast, translation of the G1 cyclin Cln3 is regulated by a leaky scanning mechanism involving a small upstream ORF (Polymenis, 1997). In this regard, it is interesting to note that the 5'-untranslated region of the Drosophila cyclin E message contains multiple upstream ORFs. Thus, the mechanisms connecting G1/S progression to cell growth may be conserved between yeast and multicellular organisms (Zhang, 2000).
Precise body and organ sizes in the adult animal are ensured by a range of signaling pathways. Rheb (Ras homolog enriched in brain), a novel, highly conserved member of the Ras superfamily of G-proteins, promotes cell growth. Overexpression of Rheb in the developing fly causes dramatic overgrowth of multiple tissues: in the wing, this is due to an increase in cell size; in cultured cells, Rheb overexpression results in accumulation of cells in S phase and an increase in cell size. Rheb is required in the whole organism for viability (growth) and for the growth of individual cells. Inhibition of Rheb activity in cultured cells results in their arrest in G1 and a reduction in size. These data demonstrate that Rheb is required for both cell growth (increase in mass) and cell cycle progression; one explanation for this dual role would be that Rheb promotes cell cycle progression by affecting cell growth. Consistent with this interpretation, flies with reduced Rheb activity are hypersensitive to rapamycin, an inhibitor of the growth regulator target of rapamycin(TOR), a kinase required for growth factor-dependent phosphorylation of ribosomal S6 kinase (S6K). In cultured cells, the effect of overexpressing Rheb was blocked by the addition of rapamycin. These results imply that Rheb is involved in TOR signaling (Patel, 2003). Additional studies show that Rheb functions downstream of the tumor suppressors Tsc1 (tuberous sclerosis 1)-Tsc2, with Tsc2 functioning as a GAP for Rheb (Saucedo, 2003; Zhang, 2003), and that a major effector of Rheb function in controlling growth is, in fact, ribosomal S6 kinase (Stocker, 2003). It is still not clear, however, how Rheb signals to TOR (Zhang, 2003).
Given the similarities between Rheb and mutants in the InR and TOR signalling pathways, it is conceivable that Rheb represents a novel component of one of these growth control pathways. To test this possibility, a detailed epistasis analysis was performed. Examined first was whether the negative regulators of InR and TOR signalling (PTEN and Tsc1-Tsc2, respectively) could counteract the effects of Rheb overexpression. All overexpression experiments were performed in the eye using the GMR-Gal4 driver line. Expression of either PTEN or Tsc1-Tsc2 alone results in a very similar size reduction of the ommatidia when compared with control ommatidia. However, whereas expression of PTEN has no influence on the increase in ommatidial size caused by Rheb overexpression, co-expression of Tsc1-Tsc2 results in ommatidia of approximately wild-type size, indicating that the activities of Rheb and Tsc1-Tsc2 can counteract each other. Next, the enlarged ommatidia phenotype of GMR-Rheb was assayed in a number of mutant backgrounds. Reducing the activity of Drosophila protein kinase B (PKB) has no effect on ommatidial size. Similar results were obtained with hypomorphic mutations in InR and Dp110, respectively. In contrast, ommatidial size is dominantly reduced by a mutation in TOR (TOR2L1), and a suppression to wild-type size is observed in a S6K mutant background. Thus, the Rheb overexpression phenotype is dependent on TOR and S6K function, but is independent of InR signal strength. Finally, the behaviors of Rheb PTEN and Rheb Tsc1 double mutants were examined. The phenotypic consequences were assayed in mosaic animals using the ey-Flp method. As expected, the Rheb PTEN double-mutant tissue clearly displays a Rheb phenotype. The Rheb Tsc1 mutant tissue also resembles Rheb single mutants, indicating that Rheb is epistatic over (functions downstream of) Tsc1 (Stocker, 2003).
In response to starvation, eukaryotic cells recover nutrients through autophagy, a lysosomal-mediated process of cytoplasmic degradation. Autophagy is known to be inhibited by TOR signaling, but the mechanisms of autophagy regulation and its role in TOR-mediated cell growth are unclear. Signaling through TOR and its upstream regulators PI3K and Rheb is necessary and sufficient to suppress starvation-induced autophagy in the Drosophila fat body. In contrast, TOR's downstream effector S6K promotes rather than suppresses autophagy, suggesting S6K downregulation may limit autophagy during extended starvation. Despite the catabolic potential of autophagy, disruption of conserved components of the autophagic machinery, including ATG1 and ATG5, does not restore growth to TOR mutant cells. Instead, inhibition of autophagy enhances TOR mutant phenotypes, including reduced cell size, growth rate, and survival. Thus, in cells lacking TOR, autophagy plays a protective role that is dominant over its potential role as a growth suppressor (Scott, 2004).
Autophagy likely evolved in single-cell eukaryotes to provide an energy and nutrient source allowing temporary survival of starvation. In yeast, Tor1 and Tor2 act as direct links between nutrient conditions and cell metabolism. These proteins sense nutritional status by an unknown mechanism, and effect a variety of starvation responses including changes in transcriptional and translational programs, nutrient import, protein and mRNA stability, cell cycle arrest, and induction of autophagy. Autophagy thus occurs in the context of a comprehensive reorganization of cellular activities aimed at surviving low nutrient levels (Scott, 2004).
In multicellular organisms, TOR is thought to have retained its role as a nutrient sensor but has also adopted new functions in regulating and responding to growth factor signaling pathways and developmental programs. Thus in a variety of signaling, developmental, and disease contexts, TOR activity can be regulated independently of nutritional conditions. In these cases, autophagy may be induced in response to downregulation of TOR despite the presence of abundant nutrients and may potentially play an important role in suppressing cell growth rather than promoting survival. Identification of the tumor suppressors PTEN, and TSC1 and TSC2 as positive regulators of autophagy provides correlative evidence supporting such a role for autophagy in growth control. Alternatively, since TOR activity is required for proper expression and localization of a number of nutrient transporters, inactivation of TOR may lead to reduced intracellular nutrient levels, and autophagy may therefore be required under these conditions to provide the nutrients and energy necessary for normal cell metabolism and survival (Scott, 2004).
The results presented here provide genetic evidence that under conditions of low TOR signaling, autophagy functions primarily to promote normal cell function and survival, rather than to suppress cell growth. This conclusion is based on the finding that genetic disruption of autophagy does not restore growth to cells lacking TOR, but instead exacerbates multiple TOR mutant phenotypes. It is important to note that mutations in TOR do not disrupt larval feeding, and thus disruption of autophagy is detrimental in TOR mutants despite the presence of ample extracellular nutrients. The finding that autophagy is critical in cells lacking TOR further supports earlier studies suggesting that inactivation of TOR causes defects in nutrient import, resulting in an intracellular state of pseudo-starvation (Scott, 2004).
Can the further reduction in growth of TOR mutant cells upon disruption of autophagy be reconciled with the potential catabolic effects of autophagy? TOR regulates the bidirectional flow of nutrients between protein synthesis and degradation through effects on nutrient import, autophagy, and ribosome biogenesis. When TOR is inactivated, rates of nutrient import and protein synthesis decrease, resulting in a commensurate reduction in mass accumulation and cell growth. In addition, autophagy is induced to maintain intracellular nutrient and energy levels sufficient for normal cell metabolism. When autophagy is experimentally inhibited in cells lacking TOR, this reserve source of nutrients is blocked, leading to a further decrease in energy levels, protein synthesis, and growth. It is noted that autophagy may have additional functions in cells with depressed TOR signaling, including recycling of organelles damaged by the absence of TOR activity, or selective degradation of cell growth regulators, analogous to the regulatory roles of ubiquitin-mediated degradation (Scott, 2004).
Autophagy is required for normal developmental responses to inactivation of insulin/PI3K signaling in the nematode C. elegans. In response to starvation or disruption of insulin/PI3K signaling, C. elegans larvae enter a dormant state called the dauer. Autophagy has been observed in C. elegans larvae undergoing dauer formation: disruption of a number of ATG homologs interfers with normal dauer morphogenesis. Importantly, simultaneous disruption of insulin/PI3K signaling and autophagy genes results in lethality, similar to the results presented in this study. Thus despite significant differences in developmental strategies for surviving nutrient deprivation, autophagy plays an essential role in the starvation responses of yeast, flies, and worms (Scott, 2004).
The prevailing view that S6K acts to suppress autophagy was founded on correlations between induction of autophagy and dephosphorylation of rpS6 in response to amino acid deprivation or rapamycin treatment. However, the genetic data presented in this study argue strongly against a role for S6K in suppressing autophagy: unlike other positive components of the TOR pathway, null mutations in S6K do not induce autophagy in fed animals. It is suggested that the observed correlation between S6K activity and suppression of autophagy is due to common but independent regulation of S6K and autophagy by TOR. Thus, autophagy suppression and S6K-dependent functions such as ribosome biogenesis represent distinct outputs of TOR signaling (Scott, 2004).
How might TOR signal to the autophagic machinery, if not through S6K? In yeast, this is accomplished in part through regulation of Atg1 kinase activity and ATG8 gene expression (Kamada, 2000 and Kirisako, 1999). The demonstration of a role for Drosophila ATG1 and ATG8 homologs [see TG8a (CG32672) and ATG8b (CG12334)] in starvation-induced autophagy, and the genetic interaction observed between ATG1 and TOR, are consistent with a related mode of regulation in higher eukaryotes. However, it is noted that other components of the yeast Atg1 complex such as Atg17 and Atg13, whose phosphorylation state is rapamycin sensitive, do not have clear homologs in metazoans, indicating that differences in regulation of autophagy by TOR are likely (Scott, 2004).
In addition to excluding a role for S6K in suppression of autophagy, these results reveal a positive role for S6K in induction of autophagy. S6K may promote autophagy directly, through activation of the autophagy machinery, or indirectly through its effects on protein synthesis. The latter possibility is consistent with previous reports that protein synthesis is required for expansion and maturation of autophagosomes. Interestingly, despite being required for autophagy, S6K is downregulated under conditions that induce it, including chronic starvation and TOR inactivation. Consistent with this, it was found that lysotracker staining is significantly weaker in chronically starved animals or in TOR mutants than in wild-type animals starved 3-4 hr. Furthermore, expression of constitutively activated S6K has no effect in wild-type, but restores lysotracker staining in TOR mutants to levels similar to those of acutely starved wild-type animals. It is suggested that downregulation of S6K may limit rates of autophagy under conditions of extended starvation or TOR inactivation and that this may protect cells from the potentially damaging effects of unrestrained autophagy (Scott, 2004).
Co-culture and conditioned media experiments have shown that the Drosophila fat body is a source of diffusible mitogens. The fat body has also been shown to act as a nutrient sensor through a TOR-dependent mechanism and to regulate organismal growth through effects on insulin/PI3K signaling. The results in this study extend these findings by showing that this endocrine response is accompanied by the regulated release of nutrients through autophagic degradation of fat body cytoplasm. Preventing this reallocation of resources, either through constitutive activation of PI3K or through inactivation of ATG genes, results in profound nutrient sensitivity. Thus, in response to nutrient limitation, the fat body is capable of simultaneously restricting growth of peripheral tissues through downregulation of insulin/PI3K signaling and providing these tissues with a buffering source of nutrients necessary for survival through autophagy (Scott, 2004).
Reducing insulin/IGF signaling allows for organismal survival during periods of inhospitable conditions by regulating the diapause state, whereby the organism stockpiles lipids, reduces fertility, increases stress resistance, and has an increased lifespan. The Target of Rapamycin (TOR) responds to changes in growth factors, amino acids, oxygen tension, and energy status; however, it is unclear how TOR contributes to physiological homeostasis and disease conditions. This study shows that reducing the function of Drosophila TOR results in decreased lipid stores and glucose levels. Importantly, this reduction of TOR activity blocks the insulin resistance and metabolic syndrome phenotypes associated with increased activity of the insulin responsive transcription factor, FOXO. Reduction in TOR function also protects against age-dependent decline in heart function and increases longevity. Thus, the regulation of TOR activity may be an ancient 'systems biological' means of regulating metabolism and senescence, that has important evolutionary, physiological, and clinical implications (Luong, 2006).
The major cause of metabolic syndrome (defined as a cluster of metabolic abnormalities such as elevated glucose and lipid levels, related to a state of insulin resistance) and diabetes in humans is reduction of insulin signaling, but the underlying pathways and mechanisms are not completely understood. Likewise, caloric excess can lead to nutrient toxicity and desensitization of insulin signaling. Thus, dysregulation of energy homeostasis can lead to metabolic disturbances and predisposition to a variety of endocrine diseases including diabetes, cardiovascular disease, and cancer (Luong, 2006).
One major system that regulates energy homeostasis in higher metazoa is the insulin/IGF pathway. The functionally conserved components of the insulin/IGF pathway like insulin, the insulin receptor (InR), insulin receptor substrate (IRS), phosphoinositide 3-kinase (PI3K), protein kinase B (PKB, a.k.a. Akt) and the forkhead transcription factor FOXO have been shown to be involved in glucose and lipid homeostasis. Loss of insulin signaling in the periphery and in pancreatic β cells can lead to hyperglycemia and diabetes. For example, disruption of the InsR gene in the pancreatic β cells reduces islet size and insulin secretion. IRS1 knockout mice are hyperglycemic, but their pancreatic β cells hypertrophy to compensate for increased peripheral insulin resistance. In contrast, IRS2 knockout mice are diabetic because their pancreatic β cells are absent due to increased cell death. Additionally, systemic loss of insulin signaling in metazoans leads to elevated lipids as seen in the Daf-2 mutant worms, Chico/IRS mutant flies, and IRS2 ablated mice (Luong, 2006 and references therein).
Many of these insulin/IGF-mediated metabolic effects depend on the winged helix transcription factor, FOXO. FOXO was first identified in the worm, C. elegans as Daf-16, a mutation that can suppress the increased lipid levels and longevity caused by loss of Daf-2, the worm InR ortholog. There is a single evolutionarily conserved Drosophila FOXO ortholog and three mammalian FOXO genes (FOXO1, FOXO3a, and FOXO4). FOXO1 controls glucose homeostasis in both peripheral tissues and pancreatic β cells. For example, expression of a constitutively activated FOXO1 (resistant to insulin/IGF-mediated inactivation) in liver and pancreatic β cells causes hepatic insulin resistance and loss of pancreatic β cells via increased apoptosis, whereas reduction of FOXO1 function can reverse the loss of pancreatic β cells and hyperglycemia seen in the IRS2 ablated mice. Thus, FOXO is a critical mediator of insulin signaling in both insulin sending and receiving tissues (Luong, 2006).
The Tuberous Sclerosis Complex (TSC1-2)/Target of Rapamycin (TOR) pathway responds to changes in insulin/IGF levels, amino acid levels, energy charge, lipid status, mitochondrial metabolites, and oxygen tension by adjusting cell growth. In addition to its well-defined role in controlling cell growth, the TSC1-2/TOR pathway may also potentially be a critical regulator of glucose and lipid homeostasis as TSC1-2/TOR signaling functionally interacts with the insulin/IGF pathway. A role for TOR signaling in glucose and lipid homeostasis in mammalian systems is demonstrated by the S6K1 knockout mice. These mice are hyperglycemic caused by diminished insulin secretion due to reduced pancreatic β cell mass. This result is in keeping with studies that show that rapamycin treatment leads to decreased levels of translation, growth, and survival of pancreatic. However, the mS6K1 mutant mice have low lipid levels because of adipocytes that have increased fatty acid β-oxidation. Additionally, the mS6K1 mutant mice show enhanced glucose uptake upon exogenous insulin addition due to insulin hypersensitivity in peripheral tissues via loss of a negative feedback loop on IRS. Thus, TOR signaling via S6K can modulate insulin sensitivity by altering Ser307 and Ser636/639 phosphorylation and IRS protein levels (Luong, 2006).
There are additional levels where TSC1-2/TOR signaling may positively and negatively regulate insulin signaling. There are data that suggest that the IRS Ser302 site is required for signaling to TOR and S6K. Thus, ser/thr phosphorylation of the IRS proteins may mediate both positive and negative signals for energy homeostasis. Furthermore, Akt/PKB activity may also be directly regulated by the nutrient-sensitive TOR pathway. Although the insulin/IGF pathway can signal to the TSC1-2/TOR pathway, recent evidence suggests that TOR may directly control Akt/PKB function because Akt/PKB activation depends on TORC2 complex-specific TOR Ser473 phosphorylation of Akt/PKB. Thus, these studies suggest that dysregulation of TSC1-2/TOR signaling may contribute to the pathological progression of metabolic syndrome and diabetes, yet the direct role and function of TOR is unclear in this context (Luong, 2006).
Another functionally conserved energy homeostatic pathway is the AMP-activated protein kinase (AMPK) pathway (see AMP-activated protein kinase). This pathway responds to altered energy states caused by cellular stresses like mitochondrial dysfunction, anti-diabetic drugs, and exercise. Activation of the energy sensing AMPK pathway by activated AMPK as well as metformin or AICAR treatment results in decreased lipogenesis and gluconeogenesis via both central. Activated AMPK can phosphorylate TSC2, which inhibits TOR signaling, while loss of AMPK activity causes an increase in TOR signaling. However, the requirement of TOR function for the AMPK energy response is not known. These effects may also be mediated by targets including glycogen synthase, hormone-sensitive lipase, acetyl-CoA carboxylase-2, HMG-CoA reductase, p300, and p53; the different roles of these proteins in the AMPK-mediated low energy response are not well known. Furthermore, activation of AMPK leads to IRS Ser-789 phosphorylation and enhancement of insulin signaling, which suggests that the AMPK response can act separately from the TOR pathway to enhance insulin signaling. Clearly, there is a great need to understand the regulation of TSC1-2/TOR signaling as it relates to the maintenance of energy homeostasis because TOR function is implicated in both insulin/IGF and AMPK signaling (Luong, 2006).
Although TOR occupies a central node that governs catabolic or anabolic responses to different nutritional and energy states, the resultant metabolic effects of altering TOR function in a metazoan are incompletely and poorly understood. This study examines in detail the function of Drosophila TOR in terms of energy homeostasis and senescent responses. Reduction of TOR function is show to result in decreased glucose and lipid levels with concomitant increase of DILP2 from the insulin producing cells. A reduction of TOR function can block activated FOXO-mediated insulin resistance and metabolic syndrome phenotypes. Taken together, these data indicate that TOR function is required for the maintenance of energy homeostasis and organismal senescence. The additional ramifications of this study are that reduction of TOR function may have clinical utility for treating metabolic syndrome and insulin resistance (Luong, 2006).
In contrast to the elevated lipid levels caused by reduction of systemic insulin signaling, the dTOR7/P mutant (containing a P-element insert into TOR) does not show increased lipid levels. Instead, the dTOR7/P mutant shows decreased lipid levels of the fat body that depend on the function of a lipase involved in lipid metabolism. Elevated ketone bodies were observed in the hypoglycemic dTOR7/P mutant, which is indicative of the increased utilization of lipids. Studies in mammalian cardiac tissue have shown that ketone bodies provide their high energy electrons directly to complex I, the NADH dehydrogenase multienzyme complex, of the mitochondrial electron transport chain. Thus, the altered lipid levels show that TOR has a critical role in determining the fate of fats (Luong, 2006).
It has also been shown that 4EBP is involved in lipid metabolism because the increased lipid levels caused by rapamycin treatment are blocked by a 4EBP mutant. Furthermore, loss of the melted mutant has lower lipid levels, due to lowered triglyceride production. This effect is due to increased 4EBP protein levels via FOXO activation in the fatbody. However, there is no change in glucose levels. In this respect, the melted mutant resembles the FIRKO mouse because it shows decreased triglyceride levels without a change in glucose levels. The dTOR7/P mutant has a different lipid phenotype than the one caused by rapamycin treatment, which suggests that rapamycin alters TOR function in a different manner than the dTOR7/P mutant. Additionally, a novel hypomorphic dTOR FAT domain allele in combination with the dTORP allele also shows low glucose and lipid levels, which suggests that partial reduction of TOR activity represents a unique phenotypic class of TOR metabolic effects versus an allele specific phenotype. Although rapamycin affects TORC1 directly, TORC2 may be altered indirectly via TOR depletion and blocking of TORC2 assembly. Thus, it is not currently clear if the effects of rapamycin are due to inhibiting TORC1 and/or TORC2. It is known that rapamycin can impair pancreatic β cell function because it causes decreased growth and survival. Thus, rapamycin treatment can lead to elevated glucose and lipid levels, possibly as a result of systemic insulin loss (Luong, 2006).
Evidence shows that DILP2 levels are increased in the IPCs of the dTOR7/P mutant and the dTOR7/P mutant has lowered glucose levels. Thus, reduction of TOR function can lead to increased DILP2 levels and a reduction of glucose levels. Recent studies with the Drosophila miRNA-278 mutant also showed elevated DILP levels, yet displayed fatbody-mediated insulin resistance as shown by elevated d4EBP and glucose levels. It is believed that the dTOR7/P mutant represents an insulin-sensitized state because the dTOR7/P mutant shows decreased levels of the insulin resistance marker 4EBP, the dTOR7/P mutant shows decreased glucose levels, and, as discussed below, the dTOR7/P mutant blocks activated FOXO-mediated insulin resistance phenotypes. Thus, the dTOR mutant phenotype resembles a whole-animal 'insulin-sensitized' state that can function below the level of constitutive FOXO activity (Luong, 2006).
Overexpression of activated FOXO in peripheral and IPC tissues results in elevated glucose and lipid levels. Although TOR signaling can alter insulin signaling upstream of FOXO, reducing TOR function is able to reverse these effects. Thus, the results show that reduction of TOR activity can block the activated FOXO-mediated insulin resistance and metabolic syndrome phenotypes. These results suggest that strategies to dampen, reduce, or block TOR signaling may be able to overcome insulin resistance (i.e., hyperglycemia and hypertriglyceridemia) below the level of increased FOXO activity in mammalian systems (Luong, 2006).
Although FOXO has >100 potential targets that might contribute to the metabolic phenotype, this study identified Fatty acid synthase (FAS) and DILP2 as candidate mediators of the TOR effect on the FOXO metabolic phenotypes. The effect on FAS is interesting because it is upregulated by FOXO overexpression and in an IRS/chico mutant and may be an important determinant of the lipid levels. It has also been shown that activation of daf-16/FOXO can decrease the mRNA levels of a worm insulin gene, ins-7 . This result is consistent with results showing that DILP2 mRNA levels are decreased and reducing TOR activity can reverse this FOXO-mediated reduction of DILP2. These results might have parallels with a role for FOXO and TOR in the regulation of insulin levels in mammals (Luong, 2006).
A selective and unexpected regulation of TOR effectors is also seen: loss of 4EBP protein and a mild effect on S6K Ser389 phosphorylation. It has been recently shown that the 4EBP gene is a target of FOXO in Drosophila and thus may represent one of the TOR targets responsible for contributing to the FOXO-mediated metabolic phenotypes. It has also been shown that daf-15/Raptor is a target of daf-16/FOXO in C. elegans and may also contribute to the TOR metabolic and senescent phenotypes. Raptor may also account for the selective difference in the regulation of 4EBP and S6K function by TOR because Raptor binds to both S6K and 4EBP and loss of 4EBP may allow for more S6K binding to Raptor for TOR-mediated phosphorylation. Thus, these results suggest that reduction of TOR function may have selective effects on translation (Luong, 2006).
Reduction of TOR function does not provide resistance against acute stresses or cause sterility. This result is in contrast to the yeast TOR1 mutant, which shows elevated stress resistance, and the d4EBP mutant, which shows stress and starvation sensitivity. Nevertheless, the dTOR7/P mutant has an increased lifespan. This result is in keeping with the yeast, worm and fly studies that show that loss of TOR signaling can increase lifespan, as a major mediator of caloric restriction. Thus, alterations of TOR signaling contribute to the regulation of lifespan (Luong, 2006).
It is also seen that reduction of TOR activity prevents age-dependent functional decline of heart performance. It is not currently clear how TOR is regulating these organ and organismal responses, but the altered lipid metabolism may underlie these changes. For example, changes in lipid metabolism can both autonomously and non-autonomously affect heart function. Thus, reduction of TOR function may reallocate energy stores preferentially for the control of ‘long-term’ responses such as lifespan and organ maintenance. Importantly, there are many potential links between changes in energy homeostasis with alterations in aging and organ senescence. Channelling diverse stimuli like amino acids, growth factors, oxygen tension, and energy charge into the TOR pathway may be an economic method to mobilize fuel stores like lipids to counteract these fluctuations (Luong, 2006).
The conservation of basic mechanisms between Drosophila and mammals is well established. It has been shown that disruption of insulin signaling in non-mammalian systems like Drosophila results in altered glucose and lipid levels. Reducing TOR function can reverse activated FOXO-mediated insulin resistance phenotypes induced in both insulin producing and insulin receiving tissues, and thus this study provides the first direct evidence that reducing TOR function may have a clinical benefit to counter insulin resistance, metabolic syndrome, and/or diabetes. Furthermore, altering TOR signaling may underlie the benefits of various diet and nutritional regimens. These results demonstrate the utility of using the powerful genetics of this system to unravel the complex pathways involved in maintaining glucose and lipid homeostasis. In unraveling the complex genetic network of TOR and InR signaling, although far from completion, the Drosophila model has been indispensable in finding critical components and uncovering functionally important genetic interactions between these two pathways. Thus, the basic mechanisms controlling glucose and lipid homeostasis, including mechanisms by which the TSC1-2/TOR pathway influences insulin signaling as well as the influence of TSC1-2/TOR signaling on peripheral tissue and IPC physiology, are also functionally conserved (Luong, 2006).
This study has described a new use for reducing TOR activity to block insulin resistance, metabolic syndrome, and diabetic-like phenotypes downstream of activated FOXO, underlining the utility of the Drosophila model to identify and analyze components and compounds that block insulin resistance and metabolic syndrome phenotypes as well as pathological aspects of aging and organ senescence (Luong, 2006).
Determining how growth and differentiation are coordinated is key to understanding normal development, as well as disease states such as cancer, where that control is lost. Growth and neuronal differentiation are coordinated by the insulin receptor/target of rapamycin (TOR) kinase (InR/TOR) pathway. The control of growth and differentiation diverge downstream of TOR. TOR regulates growth by controlling the activity of S6 kinase (S6K) and eIF4E. Loss of s6k delays differentiation, and is epistatic to the loss of tsc2, indicating that S6K acts downstream or in parallel to TOR in differentiation as in growth. However, loss of eIF4E inhibits growth but does not affect the timing of differentiation. This study shows that there is crosstalk between the InR/TOR pathway and epidermal growth factor receptor (EGFR) signaling. InR/TOR signaling regulates the expression of several EGFR pathway components including pointedP2 (pntP2). In addition, reduction of EGFR signaling levels phenocopies inhibition of the InR/TOR pathway in the regulation of differentiation. Together these data suggest that InR/TOR signaling regulates the timing of differentiation through modulation of EGFR target genes in developing photoreceptors (McNeill, 2008).
Tight coordination of growth and differentiation is essential for normal development. InR/TOR signaling controls the timing of neuronal differentiation in the eye and leg in Drosophila. This study demonstrates that the InR/TOR pathway regulates neuronal differentiation in an S6K-dependent, but 4EBP/eIF4E-independent manner. It has previously been impossible to determine whether InR/TOR signaling was acting downstream or in parallel to the EGFR/MAPK pathway. Using argos and rho as reporters this study shows that the InR/TOR pathway is able to regulate EGFR/MAPK signaling downstream of MAPK. Moreover, pntP2 expression is up- and downregulated by activation or inhibition of InR/TOR signaling, respectively, and InR/TOR and EGFR pathways interact through pntP2. Taken together these data suggest that temporal control of differentiation by the InR/TOR pathway is achieved by modulation of EGFR pathway transcriptional targets in differentiating PRs (McNeill, 2008).
TOR is part of two multimeric complexes (TORC1 and TORC2) and is a core component of the InR pathway. TORC1 activity is regulated by nutrient and energy levels providing a conduit for hormonal and catabolic cellular inputs. Growth is regulated by two downstream targets of TORC1: S6K and 4EBP. The current data demonstrate that upstream of TORC1, differentiation and growth are regulated by the same factors. Downstream of TORC1, differentiation and growth differ significantly in that loss of s6k, but not eIF4E (or overexpression of 4EBP) affects differentiation. eIF4E regulates 7-methyl-guanosine cap-dependent translation and is the rate-limiting factor in translation initiation. The finding that eIF4E does not affect differentiation suggests that the temporal control of differentiation is not based on a translation initiation-dependent mechanism. Strikingly, loss of s6k blocks the precocious differentiation induced by loss of tsc2. Given the relatively weak effects of loss of s6k this may seem surprising. However, the degree of suppression is similar to the effect of loss of s6k on the overgrowth phenotype caused by loss of tsc2, namely, tsc2, s6k double-mutant cells are the same size as wild-type cells. Although loss of eIF4E has no affect on differentiation it may act redundantly with another factor, such as s6k. Testing this hypothesis though is technically challenging since the Drosophila genome contains eight different eIF4E isoforms. It will be interesting in future to test whether any of these isoforms regulate differentiation or alternatively whether eIF4E and s6k act redundantly. Although further work is required to determine the precise relationship between S6K and the InR/TOR pathway, the data point to a critical role of S6K in coordinating neuronal differentiation and growth (McNeill, 2008).
As in other neuronal systems, differentiation of PRs in the Drosophila eye occurs in a stereotyped manner. The advantage of the Drosophila retina as an experimental system is that the PRs differentiate spatiotemporally. Using this feature, as well as a series of cell-type-specific antibodies, this study has demonstrated that InR/TOR signaling is selective in the cell-types that it affects. The differentiation of PRs 2/5, 3/4, and 8 are unaffected by perturbations in InR/TOR signaling, whereas PRs 1, 6, and 7 and cone cells are dependent on this pathway for temporal control of differentiation. Interestingly the affected cells all differentiate after the second mitotic wave. However, regulators of the cell cycle do not affect the temporal control of differentiation. Why then are PRs 1, 6, and 7 and cone cells specifically affected? In cells with increased InR/TOR signaling, the expression of argos, rho, and pntP2 is precocious and increased throughout the clone, suggesting that the upregulation of EGFR signaling occurs in all cells. However, decreasing EGFR activity using a hypomorphic pntP2 allele specifically affects the differentiation of PRs 1, 6, and 7 and cone cells. Interestingly, pntP2 expression in differentiated cells is also restricted to PRs 1, 6, and 7 and cone cells. These observations suggest that differentiation of PRs 1, 6, and 7 and cone cells is critically dependent on EGFR levels signaling through pntP2. Therefore, although activation of InR/TOR signaling causes upregulation of EGFR transcriptional targets in all cells as they differentiate, the phenotypic effect is seen only in PRs 1, 6, and 7 and cone cells since these cells are highly sensitive to EGFR activity signaling through pntP2. This possibility is supported by the fact that precocious differentiation caused by overexpression of Dp110 can be suppressed by the simultaneous reduction of pntP2 levels. The complete suppression of the Dp110 differentiation phenotype by simultaneous reduction of pntP2 strongly suggests that pntP2 acts downstream of Dp110 and InR/TOR signaling in a pathway that regulates the temporal control of differentiation. It has been suggested that later differentiating PRs require higher levels of EGFR activity than their earlier differentiating neighbors. In particular, the activation of PR 7 requires both EGFR and Sevenless RTKs. In the case of InR/TOR pathway activation it may be that, through its regulation of EGFR downstream targets, the 'second burst' of RTK activity is enhanced causing PRs 1, 6, and 7 and cone cells to differentiate precociously. There may also be other as yet unidentified factors through which the InR/TOR pathway controls the expression of Aos and rho in PRs 2-5 and 8 (McNeill, 2008).
Activation of insulin and insulin-like growth factor receptors in mammalian systems is well known to elicit a response via the Ras/MAPK pathway. However, loss of the InR in the Drosophila eye does not result in a loss of PRs, a hallmark of the Ras pathway, nor does mutation of the putative Drk binding site in chico affect the function of the Drosophila IRS. In accordance with these data no change is seen in dpERK staining when the InR/TOR pathway is activated in the eye disc. Rather than a direct activation of Ras signaling by the InR, the data suggest that in the developing eye crosstalk between these pathways occurs at the level of regulation of the expression of EGFR transcriptional outputs. The most proximal component of the EGFR pathway that is regulated by InR/TOR signaling is pntP2. However, the data suggest that temporal control of PR differentiation requires concerted regulation of EGFR transcriptional outputs, since overexpression of pntP2 alone is not sufficient to cause precocious differentiation, whereas overexpression of activated EGFR is sufficient. Interestingly, microarray analyses of Drosophila and human cells have shown that the InR/TOR pathway regulates the expression of hundreds of genes. The mechanism by which this transcriptional control is exerted has yet to be elucidated. It will be interesting in future to determine the extent of transcriptional crosstalk between InR/TOR and EGFR pathways in developing neurons (McNeill, 2008).
Cell growth arrest and autophagy are required for autophagic cell death in Drosophila. Maintenance of growth by expression of either activated Ras, Dp110, or Akt is sufficient to inhibit autophagy and cell death in Drosophila salivary glands, but the mechanism that controls growth arrest is unknown. Although the Warts (Wts) tumor suppressor is a critical regulator of tissue growth in animals, it is not clear how this signaling pathway controls cell growth. This study shows that genes in the Wts pathway are required for salivary gland degradation and that wts mutants have defects in cell growth arrest, caspase activity, and autophagy. Expression of Atg1, a regulator of autophagy, in salivary glands is sufficient to rescue wts mutant salivary gland destruction. Surprisingly, expression of Yorkie (Yki) and Scalloped (Sd) in salivary glands fails to phenocopy wts mutants. By contrast, misexpression of the Yki target bantam is able to inhibit salivary gland cell death, even though mutations in bantam fail to suppress the wts mutant salivary gland-persistence phenotype. Significantly, wts mutant salivary glands possess altered phosphoinositide signaling, and decreased function of the class I PI3K-pathway genes chico and TOR suppressed wts defects in cell death. Although it has been shown that salivary gland degradation requires genes in the Wts pathway, this study provides the first evidence that Wts influences autophagy. These data indicate that the Wts-pathway components Yki, Sd, and bantam fail to function in salivary glands and that Wts regulates salivary gland cell death in a PI3K-dependent manner (Dutta, 2008).
Wts was identified as a protein that is expressed during autophagic cell death of Drosophila larval salivary glands with a high-throughput proteomics approach. This was surprising, given that wts RNA was not detected with DNA microarrays. Therefore, this study investigated whether Wts is present in salivary glands, and it was determined to be constitutively expressed at stages before and after the rise in ecdysone that triggers autophagic cell death. Animals that are homozygous for the hypomorphic wtsP2 allele, which is caused by a P element insertion, are defective in salivary gland cell death (Martin, 2007). Significantly two forms of Hpo are expressed during stages preceding salivary gland cell death, suggesting that phosphorylated Hpo is present in these cells and that this signaling pathway is activated (Dutta, 2008).
These studies indicate that Wts and other core components of this tumor-suppressor pathway are required for autophagic cell death of Drosophila salivary glands. wts is required for cell growth arrest and for proper regulation of caspases and autophagy, which contribute to the destruction of salivary glands. Although it is well known that cell division, cell growth, and cell death are important regulators of tissue and tumor size, it has been unclear whether a mechanistic relationship exists between cell growth and control of cell death (Dutta, 2008).
It is possible that wts and associated downstream growth-regulatory mechanisms could suppress cell death in other animals and cell types. Autophagic cell-death morphology has been reported in diverse taxa, but little is known about the mechanisms that control this form of cell death, and this lack of understanding is probably related to the limited investigation of physiologically relevant models of this process (Dutta, 2008).
This study used steroid-activated autophagic cell death of salivary glands as a system to study the relationship between cell growth and cell death. It is logical that cell growth influences cell death in salivary glands, given that autophagy is known to be regulated by class I PI3K signaling, which contributes to the death of these cells (Berry, 2007). It is unclear whether growth arrest is a determinant of autophagic cell death in other cell types and animals, and this question is important to resolve because of the importance of growth and autophagy in multiple disorders, including cancer. wts mutant salivary gland cells fail to arrest growth at the onset of puparium formation, and this suppresses the induction of autophagy. The inhibitor of apoptosis DIAP1 influences salivary gland cell death and is one of the best-characterized target genes of the Wts signaling pathway, but DIAP1 levels are not altered in wts mutant salivary glands. Significantly, the data provide the first evidence that Wts regulates autophagy and support previous studies indicating that caspases and autophagy function in an additive manner during autophagic cell death. Given the importance of both the Wts pathway and autophagy in human health, it is critical to determine whether this relationship exists in other cells (Dutta, 2008).
Cell growth and division are often considered to be synonymous, even though they are controlled by independent mechanisms. The Wts signaling pathway must influence cell growth, but most studies have emphasized the influence of this pathway on cell division and death. bantam is the only previously studied gene that is regulated by the Wts pathway and that is known to regulate cell growth. However, the mechanism of bantam action remains obscure. The current studies suggest the possibility that Wts may regulate growth via different mechanisms and that the nature of this regulation may depend on cell context. It is premature to conclude that bantam regulates a completely novel cell growth program, but the fact that misexpression of bantam stimulates cell growth in the absence of changes in a phosphoinositide marker and that chico and TOR fail to suppress the bantam-induced salivary gland-persistence phenotype minimally suggests that this microRNA regulates genes downstream of TOR. Significant progress has been made in the identification of microRNA targets, and future studies should resolve the mechanism underlying bantam regulation of cell growth (Dutta, 2008).
Recent studies of Wts signaling in Drosophila have identified a linear pathway that terminates with Yki and Sd regulation of effector genes that influence cell growth, cell division, and cell death. These studies indicate that the Wts pathway may not always regulate downstream effector genes via Yki and Sd, given that Yki expression was not able to phenocopy the wts mutant salivary gland destruction and expression of Sd induced premature degradation of salivary glands. Although bantam expression is sufficient to induce growth and inhibit cell death in salivary glands, bantam function is not required for the wts mutant phenotype. wts mutant salivary glands possess altered markers of PI3K signaling, and their defect in cell death is suppressed by chico and TOR. Combined, these results indicate that Wts regulates cell growth and cell death via a PI3K-dependent, and Yki- and Sd-independent, mechanism. Future studies will determine whether Wts regulates cell growth in a PI3K-dependent manner in other cells and animals (Dutta, 2008).
Stem cells depend on intrinsic and local factors to maintain their identity and activity, but they also sense and respond to changing external conditions. Germline stem cells (GSCs) and follicle stem cells (FSCs), the latter being somatic stem cells, in the Drosophila ovary respond to diet via insulin signals. Insulin signals directly modulate the GSC cell cycle at the G2 phase, but additional unknown dietary mediators control both G1 and G2. Target of rapamycin, or TOR, is part of a highly conserved nutrient-sensing pathway affecting growth, proliferation, survival and fertility. This study shows that optimal TOR activity maintains GSCs but does not play a major role in FSC maintenance, suggesting differential regulation of GSCs versus FSCs. TOR promotes GSC proliferation via G2 but independently of insulin signaling, and TOR is required for the proliferation, growth and survival of differentiating germ cells. TOR also controls the proliferation of FSCs but not of their differentiating progeny. Instead, TOR controls follicle cell number by promoting survival, independently of either the apoptotic or autophagic pathways. These results uncover specific TOR functions in the control of stem cells versus their differentiating progeny, and reveal parallels between Drosophila and mammalian follicle growth (LaFever, 2010).
The Drosophila ovary houses stem cells in the germarium, the anterior-most portion of each ovariole. Two or three germline stem cells (GSCs) in a specialized niche self-renew and produce cystoblasts, which divide four times with incomplete cytokinesis to form germline cysts containing one oocyte and fifteen nurse cells. Follicle stem cells (FSCs) self-renew and produce follicle cells that envelop each cyst to form an egg chamber, or follicle. After leaving the germarium, each follicle develops through fourteen stages, forming a mature oocyte. As the cyst grows, follicle cells divide mitotically until stage 7, when they begin endoreplicating. Yolk uptake or vitellogenesis initiates at stage 8. The control of distinct stem cell populations and their differentiating progeny can thus be probed in this system (LaFever, 2010).
Ovarian stem cells and their progeny respond to diet. On a protein-rich diet, GSCs and FSCs proliferate rapidly and their descendents divide and grow robustly. On a protein-poor diet, proliferation and growth are slowed, early germline cysts die and vitellogenesis is blocked. Insulin signaling is required for all responses, except early cyst viability. Insulin-like peptides directly promote GSC division, cyst growth and vitellogenesis and indirectly control GSC maintenance. Insulin-like peptides promote GSC G2 progression through PI3 kinase and FOXO; however, additional diet mediators control both G1 and G2 (LaFever, 2010 and references therein).
This study reveals specific Tor roles in Drosophila GSCs versus FSCs. Although Tor is required for proper proliferation of GSCs and FSCs, it plays a major role in GSC, but not FSC, maintenance. TOR also differentially regulates stem cells versus their progeny. Tor is necessary for early cyst proliferation, growth and survival by preventing apoptosis. By contrast, TOR does not regulate follicle cell proliferation and controls follicle cell growth and survival independently of apoptosis or autophagy. Follicle cell TOR activity also affects underlying cyst growth. Finally, TOR regulates these processes via insulin-dependent and -independent mechanisms. These studies uncover specific roles for TOR in the control of stem cells and their differentiating progeny in the Drosophila ovary. TOR is a known nutrient sensor in many systems; it is therefore speculated that TOR is part of a broadly conserved mechanism that ties stem cell maintenance and function, and the survival, proliferation and growth of their descendents, to diet-dependent factors (LaFever, 2010).
This study has reveal strikingly specific effects of Tor on GSCs, FSCs and their progeny. Coupled to studies showing the conserved role of TOR as a nutrient sensor, these results address how specific effects of a nutrient-responsive factor might contribute to the coordination between different stem cell populations and their descendents (LaFever, 2010).
Both insulin signals and TOR are nutrient-sensing factors that converge on G2 to regulate Drosophila GSC proliferation. G2 regulation in response to diet/insulin signals also occurs in Drosophila male GSCs and in Caenorhabditis elegans germline precursors. Starvation promotes deleterious mutations during Saccharomyces cerevisae division, and cancer cells form repair foci during a delayed G2 upon DNA damage. The multitude of GSC G2 regulators might reflect a mechanism to ensure genomic integrity under poor dietary conditions (LaFever, 2010).
Although TOR regulates the G1-S transition, it also modulates G2-M in S. cerevisae, Schizosaccharomyces pombe and mammalian cells. Combined with the Tor role in GSC G2, the increased phosphorylation of 4E-BP specifically during M suggests that a TOR activity increase might precede the G2-M transition. Interestingly, activated TOR is highly enriched at the mitotic spindles of rat ovarian granulosa cells and TOR inhibition by rapamycin impairs their proliferation. Marked increases in S6K activity and 4E-BP1 phosphorylation in M occur in HeLa cells, further suggesting TOR activity cell cycle regulation as part of a conserved mechanism to tie G2-M to nutrient availability (LaFever, 2010).
Although both GSCs and FSCs require Tor for normal proliferation, only GSC maintenance requires optimal TOR activity. These distinctions do not reflect a fundamental difference between germline and somatic stem cells because TOR appears to control the maintenance of several, although probably not all, mammalian somatic stem cell types. In hematopoietic stem cells, Tsc1 or PTEN loss results in increased TOR signaling and short-term expansion, but also progressive stem cell depletion. TOR activation downstream of Wnt1 overexpression leads to transiently increased hair follicle proliferation followed by stem cell loss. By contrast, PTEN mutant ovarian granulosa cells do not become depleted, despite elevated TOR activity. Because granulosa cells might derive from stem cells, it is tempting to speculate that maintenance of these stem cells might not require precise TOR regulation, similar to Drosophila FSCs (LaFever, 2010).
Ovarian stem cells and their progeny respond to TOR differently. Reduced Tor activity leads to apoptosis of 8- and 16-cell cysts, but Tor mutant GSCs do not appear to undergo apoptotic or autophagic death. The niche might conceivably prevent GSC death. Indeed, there are reports of GSC cell death within their in vivo niche. Consistent with the niche promoting GSC survival, GSCs die at higher rates when separated from somatic cells in culture. Laser ablation of the single apical niche cell causes death of Locusta migratoria male GSCs. This model, however, does not account for a normal number of Tor mutant cystoblasts and 2-cell cysts. Perhaps a combination of niche displacement and growth defects leads to Tor mutant 8- and 16-cell cyst death (LaFever, 2010).
Reduced Tor activity slows FSC proliferation, but has no effect on the cell cycle of follicle cells. This striking difference suggests that follicle cell proliferation might be largely insensitive to direct effects of diet. Follicle cells might instead receive their primary cue to divide from the underlying germline, perhaps via the actomyosin cytoskeleton. Consistent with this idea, when germline cyst growth is slowed down by InR or Myc mutation, surrounding wild-type follicle cells adjust their numbers accordingly, although it remains to be determined if this reflects changes in follicle cell proliferation per se (LaFever, 2010).
Cell competition can occur when cell populations with different growth capacities coexist. It has been proposed that a cell senses the translational capacity of their neighbors and thus distinguishes 'winner' versus 'loser' cells. The 'losers' undergo apoptosis and secrete factors that stimulate 'winner' proliferation. Although Tor regulates growth and translation, Tor mutant follicle cells do not exhibit apoptosis, but are instead extruded from mosaic monolayers. Apoptosis-independent extrusion of cells with compromised Decapentaplegic (DPP, a bone morphogenetic protein family member) signaling has been reported in mosaic Drosophila wing disc epithelia. This similarity suggests a possible connection between DPP signaling and TOR (LaFever, 2010).
TOR can be activated downstream of insulin signaling but also receives additional inputs. Insulin signaling controls germline growth via TOR, whereas insulin (via FOXO) and TOR signaling regulate GSC proliferation in parallel. Tor-null ovarian cell defects are also more severe than InR-null defects, implying that TOR receives additional inputs during oogenesis (LaFever, 2010).
Amino acid transport activates TOR signaling in Drosophila and mammals. The Drosophila genome predicts approximately 40 amino acid transporters and recent evidence suggests that methionine is a key dietary amino acid for oogenesis in Drosophila (Grandison, 2009). Further studies should investigate how various classes of amino acid transporters affect ovarian TOR signaling and amino acid requirements for specific oogenesis processes (LaFever, 2010).
4E-BP, encoded by Thor, represses cap-dependent translation via eIF4E inhibition. TOR phosphorylates and inhibits 4E-BP, leading to translation de-repression. 4E-BP, however, does not mediate Tor ovarian phenotypes, suggesting that TOR probably acts through S6K or MYC. Indeed, S6K overexpression partially restores Tor mutant growth, viability and fertility, whereas MYC loss causes germline growth phenotypes similar to Tor defects (LaFever, 2010).
Whether or not 4E-BP is required in any other tissues to mediate the effects of reduced TOR activity remains unclear. Although overexpression of eIF4E increases cell growth rates and overexpression of 4E-BP results in smaller cell size, loss of 4E-BP does not phenocopy eIF4E overexpression. Furthermore, Thor mutation has no obvious phenotype in Drosophila except for increased sensitivity to stress and impaired innate immunity. Although Thor is required for dietary restriction effects on lifespan, no reports of Tor Thor double mutants exist in the literature (LaFever, 2010).
The results bring to light interesting parallels between the role of TOR in Drosophila and mammalian ovaries. Insulin and TOR signaling are active in mammalian ovaries and rapamycin inhibits follicle growth in cultured mouse ovaries, suggesting similar regulation of oocyte growth and follicle cell numbers between Drosophila and mammals. Although adult mammalian ovaries do not contain GSCs, overexpression of either insulin or TOR signaling in mouse primordial germ cells leads to premature ovarian failure caused by the hyperactivation and subsequent depletion of the primordial germ cell pool, a phenotype that is arguably reminiscent of the rapid loss of Tsc1 mutant GSCs (LaFever, 2010).
Homeostatic mechanisms operate to stabilize synaptic function; however, little is known about how they are regulated. Exploiting Drosophila genetics, a critical role was uncovered for the target of rapamycin (TOR) in the regulation of synaptic homeostasis at the Drosophila larval neuromuscular junction. Loss of postsynaptic TOR disrupts a retrograde compensatory enhancement in neurotransmitter release that is normally triggered by a reduction in postsynaptic glutamate receptor activity. Moreover, postsynaptic overexpression of TOR or a phosphomimetic form of S6 ribosomal protein kinase, a common target of TOR, can trigger a strong retrograde increase in neurotransmitter release. Interestingly, heterozygosity for eIF4E, a critical component of the cap-binding protein complex, blocks the retrograde signal in all these cases. These findings suggest that cap-dependent translation under the control of TOR plays a critical role in establishing the activity dependent homeostatic response at the NMJ (Penney, 2012).
A growing consensus by neurobiologists suggests that a balance exists between forces that promote and those that hinder synaptic growth and function, ensuring proper synaptic connectivity and functional stability in the nervous system. A robust retrograde signaling mechanism at the Drosophila NMJ carries out the task of adjusting synaptic strength in response to a reduction in postsynaptic receptor function in GluRIIA mutants. Genetic analysis suggests that postsynaptic activity of TOR plays a key role in the ability of this retrograde signaling to carry out its function. The current findings are consistent with a model in which TOR, through activation of S6K and inhibition of 4E-BP, ensures the efficiency of cap-dependent translation in muscles and allows for the retrograde compensation to take place (Penney, 2012).
Interestingly, a moderate to strong reduction in TOR activity in the TorE161K/TorΔP mutant combination does not influence normal synaptic growth and has only a mild effect on baseline synaptic transmission. However, the findings indicate that once synaptic activity is compromised, i.e., in GluRIIA mutants, TOR becomes critical for the retrograde induction of homeostatic signaling. Furthermore, the findings suggest that TOR activity is required throughout larval development, as its inhibition by rapamycin for 12 hr during late stages of larval development is sufficient to block the retrograde signal. In addition, it was found that TOR can induce a retrograde increase in neurotransmitter release in wild-type animals, indicating that TOR can also act as an instructive force to regulate synaptic strength. These results together lead one to envision that under metabolic stress, during dietary restriction or as a result of aging perhaps, TOR could function as a modulator of neuronal function. As such, the identification of TOR as a key player in establishing retrograde signaling across synapses offers new insights into how defects in this aspect of translational regulation may underlie the destabilization of synaptic activity in neural circuits leading to abnormal neural function and behavior associated with diseases such as tuberous sclerosis complex (TSC), autism, mental retardation, and schizophrenia, where regulation of TOR activity may be altered. In fact, in animal models for TSC, hyperactivity in circuits and the susceptibility to epileptic activity can be diminished in response to rapamycin treatment. Based on the current results, it is conceivable that in TSC animals, when TOR is upregulated, synaptic activity in circuits is enhanced due to the retrograde action of TOR on neurotransmitter release, in a manner independent of growth related phenotype associated with TOR gain of function. Therefore, the results reveal a role for TOR in the retrograde regulation of neurotransmitter release in neurons, an avenue to explore aimed at potential therapeutic approaches (Penney, 2012).
Based on genetic interaction experiments and biochemical assessment, it is concluded that TOR normally acts downstream of synaptic activity. It was observed that postsynaptic phosphorylation of S6K, a bona fide TOR target, is increased in GluRIIA mutants, suggesting that TOR signaling may be upregulated in these mutants. Consistently, the genetic experiments show that removal of one gene copy of either Tor or S6k is sufficient to block the homeostatic response in GluRIIA mutants. Furthermore, when TOR is overexpressed in GluRIIA mutants no additional increase in quantal content is observed. This lack of an additive effect suggests that a common molecular pathway may be utilized by GluRIIA mutants and larvae overexpressing TOR. This is further supported by the observations that the enhancement in neurotransmission in response to TOR (or S6K) overexpression and that triggered in GluRIIA loss of function are both highly dependent on wild-type availability of eIF4E. These results together support the idea that TOR functions downstream of synaptic activity at the NMJ. Further experiments are needed to understand how changes in synaptic activity may regulate the activity of TOR (Penney, 2012).
The findings are consistent with a growing body of evidence that implicates the involvement of TOR/S6K in the regulation of synaptic plasticity in mammals. The results indicate that TOR/S6K may be exerting their function through a retrograde mechanism to enhance neurotransmission. As such, the findings reveal a novel mode of action for TOR, through which it can modulate circuit activity in higher organisms. Further experiments are required to verify if this mode of action is conserved in higher organisms (Penney, 2012).
One potential way in which general translational mechanisms can lead to specific changes in synaptic function is through localized translation. In both vertebrates and invertebrates, local postsynaptic translation is required for normal synaptic plasticity and is itself modulated by synaptic function. This is perhaps best demonstrated in cultured hippocampal neurons, where local protein synthesis at postsynaptic sites is regulated by postsynaptic activity. It appears that this modulation is, at least in part, mediated by the action of synaptic activity on the function of elongation factor, eEF2. Interestingly, blocking NMDA mediated miniatures leads to phosphorylation and inhibition of eEF2 (Sutton, 2007). The current findings are consistent with a role for miniature synaptic activity in the regulation of postsynaptic translation: in GluRIIA mutants, a reduction in miniature amplitude (and perhaps in postsynaptic calcium influx) leads to an upregulation of TOR activity as evident in the increase in S6K phosphorylation. However, at this point it cannot be conclusively show that the effect of postsynaptic TOR is localized. Further experiments are needed to verify whether these changes occur at specific postsynaptic loci at the NMJ (Penney, 2012).
The major task of the cap-binding complex is binding to the 5′UTR of the mRNA and unwinding it, so that the ribosome can interact with the mRNA and initiate translation (Ma, 2009). The results suggest that this stage of translation is the most critical for the induction of retrograde compensation. As the results suggest, once the 5′UTR is unwound, changes in the availability of the ternary complex and translation elongation are less critical for the induction of retrograde signaling. On the other hand, TOR would have a two-fold function in this scenario: one through its inhibitory action on 4E-BP, promoting eIF4Es ability to bind the 5′ cap structure, and another through its activation of S6K, which would ultimately increase the helicase ability of eIF4A to unwind mRNA 5′UTR secondary structure. This notion was supported by the results of an in vivo reporter assay showing a significant increase in translation of a reporter that bore a complex 5′UTR in response to TOR overexpression. Based on the current findings, it is speculated that perhaps genes with highly structured 5′UTRs are among the mRNAs triggered when postsynaptic activity is reduced in GluRIIA mutants or when TOR is overexpressed in muscles. The next challenge is to identify and characterize these genes, a discovery that will likely lead to a better understanding of how homeostatic mechanisms are regulated at the synapse (Penney, 2012).
Tissue-specific stem cells and their niches are organized into functional units that respond to external cues in order to maintain organ homeostasis. Insulin and Target of rapamycin (Tor) signaling mediate external cues that control adult niches and stem cells. Whether these pathways play a role in the establishment of niche-stem cell units during organogenesis has been little explored. This study shows that during larval development both Insulin-like receptor (InR) and or participate in the establishment of ovarian niches and germline stem cells (GSCs) in Drosophila. Tor and InR are required cell-autonomously for the proliferation of precursors for both somatic niches and GSCs. These pathways also promote the formation of terminal filaments (part of the somatic niche). Significantly, InR, but not Tor, signaling non-autonomously promotes primordial germ cell (PGC) differentiation. Somatic attenuation of the pathway retards PGC differentiation, whereas its activation results in their precocious differentiation. It was also shown that InR-mediated PGC differentiation is independent of somatic ecdysone signaling, but that further differentiation into cysts requires an ecdysone input. These results demonstrate that Tor and InR signaling actively participate in the formation of ovarian niches and stem cells by affecting both cell numbers and differentiation. The dual influence of Tor and InR on both somatic cells and PGCs ensures that these two cell populations develop coordinately. This work further identifies a novel step in the regulation of germ cell differentiation by demonstrating that following bag of marbles expression, cyst formation requires an additional hormonal input (Gancz, 2013).
Cell growth and proliferation in the larva require energy and metabolites. Accordingly, these processes are controlled by the InR and Tor pathways, which are sensors of the metabolic state of the organism. It was previously demonstrated that Insulin and Tor signaling promote the proliferation of germline precursors in C. elegans. This finding has been extended and it was shown that, in Drosophila, both somatic cells and PGCs require Tor and InR signaling cell-autonomously for their proliferation. This response is not limited to the larval growth period. The ovary is an active organ that maintains growing populations of cells. Accordingly, in the adult, somatic follicle cells, GSCs and germline cysts respond to nutrition by changing their proliferation rate. The cell-autonomous response of both soma and germline to InR and Tor signaling represents one mechanism by which the coordination of growth within an organ is achieved (Gancz, 2013).
In addition, this study found that the Tor and InR pathways affect PGC proliferation non-cell-autonomously. Smaller somatic ovaries correlate with reduced PGC proliferation, while overexpression of InR diverts PGCs from a proliferation to a differentiation program. Thus, coordination between somatic growth and germline division is monitored and corrected by more than one mechanism. It is as yet unclear how the state of somatic growth is communicated to the germline. The secondary signal might be a local ligand or might involve direct contact with the ICs. It was previously shown that the somatic cells of the ovary can control PGC proliferation via a feedback loop involving EGFR signaling in somatic cells and an unidentified signal that represses PGC proliferation. InR and Tor signaling might somehow affect this unknown signal (Gancz, 2013).
InR and Tor signaling are required for the differentiation of somatic intermingled cells (ICs) and terminal filament (TFs). ICs fail to integrate with PGCs when somatic cells have reduced InR or Tor signaling, suggesting these pathways affect IC behavior. Similarly, ovaries with reduced somatic InR and Tor signaling develop fewer TFs. This is consistent with previous observations that diet restriction (yeast deprivation) during the third instar results in reduced ovariole number. One explanation for this reduction is the reduction in TF precursors due to early proliferation defects. However, the strong reduction in TF numbers in chico-deficient ovaries, despite the relatively normal gonad size, suggests a specific role of InR in TF cell determination. Although InR signaling has been mostly associated with cell proliferation, a role for this pathway in neuronal cell differentiation has been described. The ovary might be another organ in which InR signaling affects cell differentiation. In the ovary, InR signaling can increase the number of cap cells by modulating Notch signaling, which is required for the establishment of this cell type. Thus, InR signaling acts at least twice in niche formation: first, it is required for TF formation, and then for cap cell establishment and maintenance (Gancz, 2013).
Activation of InR signaling in the soma initiates PGC differentiation precociously, whereas its repression postpones PGC differentiation. Combined, these results show that InR signaling is required for the maturation of the two components of the stem cell unit: the somatic niches and the PGCs that will occupy them. This coordination might be important at times when nutrient availability is limited, and niche formation is retarded. If PGCs differentiated normally, prior to the formation of protective niches, this would have resulted in full germ cell differentiation and lack of GSCs. Retarding PGC differentiation at times of limited nutrient availability allows additional time for niche formation prior to full depletion of the stem cell precursors (Gancz, 2013).
In InR-overexpressing ovaries, PGCs initiate their differentiation and express bam as early as the beginning of third instar (72 hours AEL). They then arrest their development for nearly 2 days; germline cysts form only following the normal elevation in ecdysone signaling, and fail to do so in its absence. One possibility is that somatic InR signaling is required (non-autonomously) for the initiation of bam transcription, while ecdysone initiates Bam translation, thereby transforming bam-expressing PGCs into proper cystoblasts. Alternatively, somatic InR might be sufficient for cystoblast formation, but further differentiation into germline cysts requires ecdysone signaling. This issue could not be resolved using the available anti-Bam antibody because this low-affinity reagent cannot recognize the naturally low levels of Bam protein in cystoblasts. It has previously been shown that PGCs can form cysts as early as second instar following hs-bam expression. Therefore, the requirement for somatic ecdysone signaling can be overridden by ectopic, high Bam expression (Gancz, 2013).
Irrespective of the mechanism by which somatic InR promotes PGC differentiation, the results suggest that the passage from a bam-expressing cell to a germline cyst might not be as direct as previously thought. Classical studies suggested that the major event in GSC differentiation is bam expression, and that Bam is both necessary and sufficient for GSCs to differentiate into germline cysts. However, these experiments were performed in ovaries in which the soma was WT. The current data suggest that the second signal required for cyst formation emanates from the soma. In support of this notion, somatic expression of a dominant-negative form of the Rho GTPase in adult germaria results in loss of contact between GSC daughter cells and escort cells. As a result, cystoblasts fail to differentiate into cysts and linger in the germarium. Thus, the second signal that emanates from the soma is required not only for PGC differentiation, but also continuously during adult oogenesis (Gancz, 2013).
Two hormonal pathways are required to promote PGC differentiation and the initiation of oogenesis: the ecdysone and the Insulin pathways. Both are required for proper somatic proliferation and lineage differentiation, and both act non-cell-autonomously to promote PGC differentiation. Epistasis analysis shows that both InR and ecdysone are required independently in the somatic ovary for bam expression in PGCs, and that ecdysone is additionally required to prepare the soma for its role in promoting cyst development (Gancz, 2013).
Of note, no direct link was detected between the ecdysone and InR pathways in the somatic cells of the ovary, suggesting that they act in parallel. However, the two pathways are linked systemically. In particular, Insulin and Tor signaling are required in the prothoracic gland for ecdysone synthesis. Because the timing of ecdysone release is intimately connected to the timing of niche and PGC differentiation, nutrition affects gonadogenesis in a systemic manner. Combined, these data suggest that InR signaling affects the ovarian stem cell precursors on multiple levels: cell-autonomously, non-cell-autonomously from the somatic ovarian cells, and systemically (Gancz, 2013).
Neurons and other cells display a large variation in size in an organism. Thus, a fundamental question is how growth of individual cells and their organelles is regulated. Is size scaling of individual neurons regulated post-mitotically, independent of growth of the entire CNS? Although the role of insulin/IGF-signaling (IIS) in growth of tissues and whole organisms is well established, it is not known whether it regulates the size of individual neurons. The role of IIS in the size scaling of neurons in the Drosophila CNS was studied. By targeted genetic manipulations of insulin receptor (dInR) expression in a variety of neuron types it was demonstrated that the cell size is affected only in neuroendocrine cells specified by the bHLH transcription factor DimmedD (Dimm). Several populations of Dimm-positive neurons tested displayed enlarged cell bodies after overexpression of the dInR, as well as PI3 kinase and Akt1 (protein kinase B), whereas Dimm-negative neurons did not respond to dInR manipulations. Knockdown of these components produce the opposite phenotype. Increased growth can also be induced by targeted overexpression of nutrient-dependent TOR (target of rapamycin) signaling components, such as Rheb (small GTPase), Tor and S6K (S6 kinase). After Dimm-knockdown in neuroendocrine cells manipulations of dInR expression have significantly less effects on cell size. It was also shown that dInR expression in neuroendocrine cells can be altered by up or down-regulation of Dimm. This novel dInR-regulated size scaling is seen during postembryonic development, continues in the aging adult and is diet dependent. The increase in cell size includes cell body, axon terminations, nucleus and Golgi apparatus. It is suggested that the dInR-mediated scaling of neuroendocrine cells is part of a plasticity that adapts the secretory capacity to changing physiological conditions and nutrient-dependent organismal growth (Luo, 2013).
Small GTPases act as molecular switches that alternate between GTP-bound and GDP-bound states, thereby regulating a vast array of cellular parameters, including mitochondrial activity, cell growth, cell metabolism, and cell morphology. Small GTPases are activated or inactivated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs), respectively, which regulate the GDP/GTP load of the GTPase. Hence, understanding how GEFs and GAPs are regulated is an important aspect of understanding GTPase function. Compared to the regulation of GEFs, relatively little is known about how the activity of GAPs is regulated. GAPs acting on members of the Rho and Arf GTPase superfamilies are activated via membrane recruitment, causing rearrangements in the structure of the GAPs upon membrane binding. Regulation of GAPs acting on members of the Ras superfamily of GTPases is less well understood. One such GAP is composed of the TSC1/TSC2/TBC1D7 trimeric tumor suppressor complex, which acts on the small GTPase Rheb. Although it is known that activity of this complex is regulated by phosphorylation on multiple sites, it is not yet clear how these phosphorylations affect TSC1/2 activity at the molecular level (Demetriades, 2014).
The kinase TOR complex 1 (TORC1) is a potent anabolic regulator of cellular growth and metabolism that is often hyperactivated in human cancers. To be active, TORC1 needs to bind a molecule of Rheb in the active, GTP-bound state. By inactivating Rheb, the TSC1/2 complex is therefore a critical upstream inhibitor of TORC1. The TSC1/2 complex acts as a central point of integration of almost all known inputs regulating TORC1, including cellular stresses such as low oxygen or low ATP, and various growth-promoting signals, such as PI3K, Ras, TNF, and Wnt signaling. The importance of the tuberous sclerosis complex (TSC) on TORC1 signaling and growth is highlighted by the fact that TSC2-inactivating mutations have been found in various human growth-related diseases. One other important input regulating TORC1 activity is the availability of amino acids. Whether TSC2 is also involved in regulating TORC1 in response to amino acids, however, is unclear because various studies have come to differing conclusions (Demetriades, 2014).
Unlike all the other inputs that regulate TORC1 via TSC1/2 and Rheb, amino acids regulate TORC1 via a separate set of small GTPases, the Rag GTPases. The Rag GTPases form heterodimeric complexes consisting of RagA or RagB bound to RagC or RagD. These complexes are stably anchored to lysosomal membranes via the LAMTOR/Ragulator complex. In the presence of amino acids, the Rag dimers are in an 'active' conformation with RagA or RagB bound to GTP and RagC or RagD bound to GDP. The active Rag dimers recruit TORC1 to the lysosomal surface, where it binds Rheb to form an active holoenzyme. In the absence of amino acids, the Rag GAP complex termed GATOR1 causes the Rag dimers to switch into an inactive conformation containing GDP-bound RagA/B, thereby releasing TORC1 from the lysosomal surface. This causes TORC1 to become inactive, presumably because it no longer binds active Rheb on the lysosomal surface. Hence, TORC1 activation can currently be viewed as consisting of two aspects-the activation of Rheb in response to a plethora of regulatory inputs and the localization of TORC1 to lysosomal membranes in response to amino acids, which allows it to meet Rheb (Demetriades, 2014).
This study uncover subcellular localization as a mechanism regulating activity of the TSC1/2 GAP complex. Upon amino acid removal, TSC1/2 is recruited to lysosomes via binding to the Rag proteins, thereby bringing TSC1/2 in close proximity to its target, Rheb. This suggests that relocalization of GAPs to the vicinity of their substrates is one mechanism for their regulation. Unexpectedly, it was found that regulation of Rheb by TSC1/2 upon amino acid starvation is required for TORC1 to be released from lysosomal membranes. This suggests a 'dual anchoring' mechanism of TORC1 at the lysosome, perhaps with the Rag proteins playing a crucial role in recruiting TORC1 to the lysosomal membrane and Rheb helping to retain it there. Xells lacking TSC2 were found to be impaired in their response to amino acid starvation, failing to efficiently turn off TORC1. As a result, cells lacking TSC2 are very sensitive to amino acid starvation and die under conditions that control cells can cope with. In sum, these data indicate that the TSC1/2 complex is responsive to amino acid starvation and participates in amino acid signaling to TORC1. Hence, the TSC1/2 complex appears to play a role in regulating TORC1 in response to all regulatory inputs known to date (Demetriades, 2014).
The data presented in this study suggest a model whereby, in the presence of amino acids, mTORC1 accumulates on lysosomes due to a dual anchoring activity composed primarily of the Rag proteins but supported by Rheb. While amino acids are present, binding between the Rag proteins and TSC1/2 is low, causing the TSC1/2 complex to remain cytoplasmic. Upon amino acid removal, the Rag proteins cause mTORC1 to be released from lysosomes via two independent activities, both of which result from the Rag proteins shifting to an inactive conformation. First, the Rag proteins reduce their binding for mTORC1, thereby releasing one of the two activities tethering mTORC1 at the lysosome. Second, the Rag proteins actively recruit TSC2 to the lysosome. This allows TSC2 to act on Rheb, thereby releasing the second tethering activity keeping mTORC1 on the lysosome. In the absence of TSC2, this second activity is unaffected, causing mTORC1 to remain lysosomally localized (Demetriades, 2014).
The LAMTOR complex (composed of the p18, p14, and MP1 proteins) has been shown to serve as a docking point for mTORC1 and MEK/ERK complexes, regulating their recruitment to late endosomes/lysosomes and their activation status. These data demonstrate that integrity of the LAMTOR complex is critical for proper TSC2 subcellular localization upon amino acid withdrawal, therefore highlighting the importance of this scaffold complex for endomembrane-mediated activation/inactivation of signaling pathways (Demetriades, 2014).
The data presented in this study show that the TSC1/2 complex is part of the molecular machinery required for mTORC1 to respond properly to the absence of amino acids. The TSC1/2 complex responds to amino acid starvation by changing its subcellular localization and TSC2 is required for mTORC1 to be fully released from lysosomes and fully inactivated upon amino acid removal. That said, however, in cells lacking TSC2, there is nonetheless a clear initial drop in TORC1 activity upon amino acid removal. The remaining activity is then sustained indefinitely. Hence, mTORC1 appears to consist of two pools or two degrees of activation, one of which requires TSC2 to become inactive upon amino acid withdrawal and one of which responds independently of TSC2. This might explain why previous studies arrived at differing interpretations of their data because there is some response of TORC1 to amino acid removal in TSC2 null cells; however, the response is severely blunted compared to controls. Further work will hopefully shed light on these two pools of activity. Although the impairment in mTORC1 response to amino acids in TSC2 null cells is partial, it is nonetheless of critical physiological relevance because TSC2 null MEFs die upon amino acid removal in sharp contrast to control MEFs (Demetriades, 2014).
Various insights can be derived from these data. (1) Regulation of mTORC1 activation could previously be rationalized as consisting of two independent, parallel steps: first, regulation of Rheb via TSC1/2 in response to a plethora of signals including stresses and growth factor signaling, and second, regulation of mTORC1 subcellular localization to lysosomal membranes in response to amino acids. Only when mTORC1 is properly localized to meet active Rheb would an active holoenzyme form. The data presented in this study blur the distinction between these two steps because Rheb also affects mTORC1 localization, and amino acids also signal through TSC1/2. Instead, the two sets of regulatory inputs into mTORC1 appear to be more integrated. (2) Amino acid removal is 'dominant' over growth factor signaling, causing mTORC1 to shut off despite the presence of growth factors. This was previously explained by the fact that, in the absence of amino acids, mTORC1 could not localize near active Rheb to form an active complex. The dual tethering model is also consistent with this notion but for a slightly modified reason, which is that amino acid starvation acts to sever both the Rag and Rheb lysosomal tethering activities. (3) Seen from the perspective of the Rag proteins, they swap binding partners depending on the state of amino acid signaling, binding preferentially to mTORC1 in the presence of amino acids, and binding preferentially to the TSC1/2 complex in the absence of amino acids. Consequently, mTORC1 and TSC1/2 also swap subcellular localizations (Demetriades, 2014).
It was noticed that, knockdown of Rag proteins did not result in as strong a reduction in TORC1 activity in the presence of amino acids as has been previously reported. The simplest explanation is technical-that the Rag knockdowns are not strong enough to fully abrogate Rag recruitment of mTORC1 in the presence of amino acids but are sufficient to impair Rag recruitment of TSC2 in the absence of amino acids. In that case, optimizing the Rag knockdowns might lead to even stronger effects than the ones presented in this study. Two alternate biological explanations, however, might be worth investigating in the future. The first is that the data suggest a dual anchoring mechanism of mTORC1 at the lysosomal membrane-one by the Rag proteins and one by Rheb. It is possible that the relative contribution of lysosomal tethering of mTORC1 by the Rag proteins and by Rheb might depend on their relative levels of expression and activation in the system being studied. This balance will likely depend on the cell line and on cell culture conditions. A second possible explanation could be one of biological kinetics, influenced by treatment strategy. The outcome might be quantitatively different if one looks at acute amino acid removal from cells adapted to complete medium (which is done in this study) or if one looks at amino acid add-back to cells that have equilibrated their signaling to the absence of amino acids. Indeed, it was seen that, the Rag proteins were knocked down in HEK293FT cells, no dramatic reduction is seen in mTORC1 activity in the presence of amino acids. However, if amino acids are removed for 1 hr and then amino acids were re-added for 30 min, the same degree of Rag knockdown causes an obvious reduction in mTORC1 activity. Likewise, in Drosophila S2 cells, RagC knockdown only had a mild effect on TORC1 activity in untreated cells but severely blunted the ability of cells to respond to amino acid add-back. This difference between amino acid removal and amino acid add-back raises the interesting possibility that the Rag proteins are key in recruiting mTORC1 to the lysosome, a process that happens upon amino acid readdition, and that both the Rag proteins and Rheb work together to keep mTORC1 on the lysosome once it is there. Indeed, in agreement with this model, mTOR is able to be recruited to the lysosome upon amino acid readdition in TSC2 null MEFs in which Rheb is knocked down, indicating that, although Rheb tethers mTOR to the lysosome upon amino acid removal, it is not required for de novo recruitment of mTOR to the lysosome upon amino acid add-back (Demetriades, 2014).
Previous reports have shown that hyperactive mTORC1 signaling or dysregulated translation can lead to a metabolic mismatch in supply and demand, leading to cellular or organismal death. It is hypothesized that, if TSC2 is required for mTORC1 activity to respond to amino acid starvation, then TSC2 might also be necessary for cells to respond physiologically to this stress. Indeed, TSC2 knockout MEFs die upon removal of amino acids, whereas control cells do not. The fact that cells with elevated mTORC1 activity due to impaired nutrient sensing die when deprived of amino acids raises the interesting hypothesis that limiting nutrient supply to tumors of certain genotypes might have a beneficial effect on their treatment. Consistent with this effect being due to elevated mTORC1 activity, the death of TSC2 knockout MEFs is rescued by rapamycin treatment. This leads to the unexpected finding that rapamycin can actually promote cell survival under nutrient deprivation conditions, which might have therapeutic implications in mTOR-related malignancies (Demetriades, 2014).
This study identified the subcellular localization of TSC1/2 as one mechanism regulating this GAP holoenzyme. In an accompanying manuscript, It has been shown that TSC2 subcellular localization is also regulated by insulin signaling. They show that, in the absence of fetal bovine serum (FBS), TSC2 is lysosomally localized. This study shows that, in the absence of amino acids, TSC2 is lysosomally localized, even in the presence of growth factor signaling (FBS). Hence, the presence of both amino acids and growth factor signaling are required to keep TSC2 in the cytoplasm, and as long as one of the two is missing, TSC2 becomes lysosomally localized. Combined, these findings raise the possibility that TSC2 subcellular localization is a general mechanism for regulating this complex. It would be interesting to study whether the other inputs known to regulate TSC2 also affect its subcellular localization (Demetriades, 2014).
In single-cell eukaryotes the pathways that monitor nutrient availability are central to initiating the meiotic program and gametogenesis. In Saccharomyces cerevisiae an essential step in the transition to the meiotic cycle is the down-regulation of the nutrient-sensitive target of rapamycin complex 1 (TORC1; see Drosophila Tor pathway) by the increased minichromosome loss 1/ GTPase-activating proteins toward Rags 1 (Iml1/GATOR1)
In yeast, the inhibition of the nutrient-sensitive target of rapamycin complex 1 (TORC1) in response to amino acid limitation is essential for cells to transit from the mitotic cycle to the meiotic cycle. In response to amino acid starvation, the Iml1 complex, comprising the Iml1, Nitrogen permease regulator-like 2 (Npr2), and Nitrogen permease regulator-like 3 (Npr3) proteins in yeast and the respective orthologs DEPDC5, Nprl2, and Nprl3 in mammals, inhibits TORC1 activity. The Iml1 complex, which has been renamed the 'GTPase-activating proteins toward Rags 1' (GATOR1) complex in higher eukaryotes, functions as a GTPase-activating protein complex that inactivates RagsA/B or Gtr1 in mammals and yeast, respectively, thus preventing the activation of TORC1. In the yeast Saccharomyces cerevisiae, mutations in the Iml1 complex components Npr2 and Npr3 result in a failure to down-regulate TORC1 activity in response to amino acid starvation and block meiosis and sporulation. As is observed in yeast, in Drosophila, Nprl2 and Nprl3 mediate a critical response to amino acid starvation (Wei, 2014a). However, their roles in meiosis and gametogenesis remain unexplored (Wei, 2014b).
Recent reports indicate that the Iml1, Npr2, and Npr3 proteins are components of a large multiprotein complex originally named the 'Seh1-associated' (SEA) complex in budding yeast and the 'GATOR' complex in higher eukaryotes. The SEA/GATOR complex contains eight highly conserved proteins. The three proteins described above, Iml1/DEPDC5, Npr2/Nprl2, and Npr3/Nprl3, form the Iml1/GATOR1 complex and inhibit TORC1. The five remaining proteins in the complex, Seh1, Sec13, Sea4/Mio, Sea2/WDR24, and Sea3/WDR59, which have been designated the 'GATOR2' complex in multicellular organisms, oppose the activity of Iml1/GATOR1 and thus promote TORC1 activity (Wei, 2014b).
Little is known about the physiological and/or developmental requirements for the GATOR2 complex in multicellular organisms. However, in Drosophila the GATOR2 components Mio and Seh1 interact physically and genetically and exhibit strikingly similar ovarian phenotypes, with null mutations in both genes resulting in female sterility (Senger, 2011; Wei, 2014a). In Drosophila females, oocyte development takes place within the context of an interconnected germline syncytium, also referred to as an 'ovarian cyst'. Ovarian cyst formation begins at the tip of the germarium when a cystoblast, the daughter of a germline stem cell, undergoes four synchronous divisions with incomplete cytokinesis to produce 16 interconnected cells. Actin-stabilized cleavage furrows, called 'ring canals', connect cells within the cyst. Each 16-cell cyst develops with a single oocyte and 15 polyploid nurse cells which ultimately are encapsulated by a somatically derived layer of follicle cells to produce an egg chamber. Each ovary is comprised of ∼15 ovarioles that consist of a single germarium followed by a string of egg chambers in successively older stages of development. In mio- and seh1-mutant egg chambers, the oocyte enters the meiotic cycle, but as oogenesis proceeds, the oocyte fate and the meiotic cycle are not maintained stably (Senger, 2011; Wei, 2014a). Ultimately, a large fraction of mio and seh1 oocytes enter the endocycle and develop as polyploid nurse cells. A mechanistic understanding of how mio and seh1 influence meiotic progression and oocyte fate has remained elusive (Wei, 2014b).
This study demonstrates that the Iml1/GATOR1 complex down-regulates TORC1 activity to promote the mitotic/meiotic transition in Drosophila ovarian cysts. Depleting iml1 in the female germ line delays the mitotic/meiotic transition, so that ovarian cysts undergo an extra mitotic division. Conversely, mutations in Tor result in premature meiotic entry before the completion of the four mitotic divisions. Moreover, it was demonstrated that in the female germ line, the GATOR2 components Mio and Seh1 are required to oppose the TORC1 inhibitory activity of the Iml1/GATOR1 complex to prevent the constitutive down-regulation of TORC1 activity in later stages of oogenesis. These studies represent the first examination of the regulatory relationship between Iml1/GATOR1 and GATOR2 components within the context of a multicellular animal. Finally, these data reveal a surprising tissue-specific requirement for the GATOR2 complex in multicellular organisms and suggest a conserved role for the SEA/GATOR complex in the regulation of TORC1 activity during gametogenesis (Wei, 2014b).
Previous work demonstrated that in Drosophila the Iml1/GATOR1 complex mediates an adaptive response to amino acid starvation. This study tested the hypothesis that the Iml1/GATOR1 complex also has retained a role in the regulation of the early events of gametogenesis. Consistent with this model, this study found that in germline knockdowns of iml1, ovarian cysts delay meiotic entry and undergo a fifth mitotic division. This meiotic delay can be suppressed with the TORC1 inhibitor rapamycin. Thus, during Drosophila oogenesis the Iml1/GATOR1 complex promotes the transition from the mitotic cycle to the meiotic cycle through the down-regulation of the metabolic regulator TORC1. Increasing TORC1 activity by disabling its inhibitor delays meiotic progression, whereas germline clones of a Tor-null allele enter meiosis prematurely. Taken together, these data indicate that the level of TORC1 activity contributes to the timing of the mitotic/meiotic switch in Drosophila females and suggest that low TORC1 activity may be a conserved feature of early meiosis in many eukaryotes (Wei, 2014b).
However, in Drosophila, meiotic entry is not contingent on amino acid limitation at the organismal level. Indeed, the energy-intensive process of Drosophila oogenesis is curtailed dramatically when females do not have access to a protein source. Thus, to promote meiotic entry, Drosophila females must activate the Iml1/GATOR1 complex in a tissue-specific manner, using a mechanism that is independent of the overall nutrient status of the animal. At least two models can explain how Drosophila females might activate the Iml1/GATOR1 complex specifically in the germ line. In the first model, ovarian cysts locally experience low levels of amino acids during the mitotic cyst divisions and/or at the point of meiotic entry. These low levels of amino acids could reflect a non–cell-autonomous effect: The somatically derived escort cells that surround dividing ovarian cysts may function to create a low amino acid environment that triggers the activation of the Iml1/GATOR1 complex within developing ovarian cysts. Alternatively, the effect may be cell autonomous: The germ cells within dividing ovarian cysts may have a reduced ability to sense and/or import amino acids. In a second model, a developmental signaling pathway that is completely independent of local or whole-animal amino acid status directly activates the Iml1/GATOR1 complex. The identification of the upstream requirements for Iml1/GATOR1 activation in the female germ line will help distinguish between these two models (Wei, 2014b).
Although low TORC1 activity is required during early ovarian cyst development to promote the mitotic/meiotic switch, the dramatic growth of egg chambers later in oogenesis is a metabolically expensive process that is predicted to require high TORC1 activity. The current data indicate that the GATOR2 components Mio and Seh1 function to oppose the TORC1-inhibitory activity of the GATOR1 complex in the female germ line. In mio and seh1 mutants, TORC1 activity is constitutively repressed in the germ line of developing egg chambers, resulting in the activation of catabolic metabolism and the blocking of meiotic progression and oocyte development and growth (Wei, 2014b).
Previous data indicate that Mio and Seh1 act very early in oogenesis soon after the formation of the 16-cell cyst. The mio and seh1 ovarian phenotypes can be rescued by depleting the GATOR1 components nprl2, nprl3, or iml1 in the female germ line or by raising baseline levels of TORC1 activity by disabling an alternative TORC1 inhibitory complex, TSC1/2. These data are consistent with the model that the failure to maintain the meiotic cycle and the oocyte fate in mio and seh1 mutants is a direct result of inappropriately low TORC1 activity in the female germ line brought on by the deregulation of the Iml1/GATOR1 complex (Wei, 2014b).
Notably, null alleles of both mio and seh1 are viable, with many somatic tissues exhibiting no apparent developmental abnormalities and only limited reductions in cell growth. Thus, although Mio and Seh1 are critical for the activation of TORC1 and the development of the female gamete, these proteins play a relatively small role in the development and growth of many somatic tissues under nutrient-replete conditions. Whether this small role reflects the fact that components of the Iml1/GATOR1 complex are expressed at low levels in some somatic cell types or that the complex is present but needs to be activated by a signal, such as nutrient stress or a developmental signaling pathway, remains to be elucidated (Wei, 2014b).
In the future it will be important to gain a fuller understanding of the potential environmental and developmental inputs that regulate the activity of the Iml1/GATOR1 and GATOR2 complexes in multicellular organisms. These studies will provide much-needed insight into the basic mechanisms by which both environmental and developmental signaling pathways interface with the metabolic machinery to influence cell growth and differentiation (Wei, 2014b).
Three distinct RNA polymerases transcribe different classes of genes in the eukaryotic nucleus. RNA polymerase (Pol) III is the essential, evolutionarily conserved enzyme that generates short, non-coding RNAs, including tRNAs and 5S rRNA. The historical focus on transcription of protein-coding genes has left the roles of Pol III in organismal physiology relatively unexplored. Target of rapamycin kinase complex 1 (TORC1) regulates Pol III activity, and is also an important determinant of longevity. This raises the possibility that Pol III is involved in ageing. This study shows that Pol III limits lifespan downstream of TORC1. A reduction in Pol III extends chronological lifespan in yeast and organismal lifespan in worms and flies. Inhibiting the activity of Pol III in the gut of adult worms or flies is sufficient to extend lifespan; in flies, longevity can be achieved by Pol III inhibition specifically in intestinal stem cells. The longevity phenotype is associated with amelioration of age-related gut pathology and functional decline, dampened protein synthesis and increased tolerance of proteostatic stress. Pol III acts on lifespan downstream of TORC1, and limiting Pol III activity in the adult gut achieves the full longevity benefit of systemic TORC1 inhibition. Hence, Pol III is a pivotal mediator of this key nutrient-signalling network for longevity; the growth-promoting anabolic activity of Pol III mediates the acceleration of ageing by TORC1. The evolutionary conservation of Pol III affirms its potential as a therapeutic target (Filer, 2017).
The task of carrying out transcription in the eukaryotic nucleus is divided among RNA Pol I, II and III. This specialization is evident in the biogenesis of the translation machinery, a task that requires the co-ordinated activity of all three polymerases: Pol I generates the 45S pre-rRNA that is subsequently processed into mature rRNAs, Pol II transcribes various RNAs including mRNAs encoding ribosomal proteins, while Pol III provides the tRNAs and 5S rRNA. This costly process of generating protein synthetic capacity is tightly regulated to match the extrinsic conditions and the intrinsic need for protein synthesis by the key driver of cellular anabolism, TORC1. The central position of TORC1 in the control of fundamental cellular processes is mirrored by the notable effect of its activity on organismal physiology: following its initial discovery in worms, inhibition of TORC1 has been demonstrated to extend lifespan in all tested organisms, from yeast to mice, with beneficial effects on a range of age-related diseases and dysfunctions. TORC1 strongly activates Pol III transcription and this relationship suggests the possibility that inhibition of Pol III promotes longevity (Filer, 2017).
In Saccharomyces cerevisiae, each of the 17 Pol III subunits is encoded by an essential gene. This study generated a yeast strain in which the largest Pol III subunit (C160, encoded by RPC160, also known as RPO31) is fused to the auxin-inducible degron (AID). The fusion protein can be targeted for degradation by the ectopically expressed E3 ubiquitin ligase (OsTir) in the presence of indole-3-acetic acid (IAA) to achieve conditional inhibition of Pol III. It was confirmed that IAA treatment triggered degradation of the fusion protein, and it was observed that IAA treatment also improved the survival of the RPC160-AID strain upon prolonged culture. In addition, IAA treatment of the control strain lacking the AID fusion reduced its survival relative to both the same strain in the absence of IAA and to the RPC160-AID strain in the presence of IAA. Hence, Pol III depletion appears to extend the chronological lifespan in yeast. While IAA had no substantial effect on the survival of a strain carrying the AID domain fused to the largest subunit of Pol II (RPB220, also known as RP021), this strain appeared to survive better than the control strain did in the presence of IAA, indicating that inhibition of Pol II may also extend chronological lifespan. Chronological lifespan of yeast is a measure of survival in a nutritionally limited, quiescent population, whereas replicative lifespan measures the number of daughters produced by a single mother cell in its lifetime. No evidence was found that inhibition of Pol III causes an increase in the replicative lifespan in yeast (Filer, 2017).
The observed increase in chronological lifespan may simply indicate increased stress resistance and hence be of limited relevance to organismal ageing. To examine the role of Pol III in organismal ageing directly, animal models were examined. RNA-mediated interference (RNAi) was initiated against rpc-1, the Caenorhabditis elegans orthologue of RPC160, in worms from the L4 stage, causing a partial knockdown of rpc-1 mRNA. This consistently extended the lifespan of worms at both 20°C and 25°C. To reduce Pol III activity in Drosophila melanogaster, a P-element insertion that deletes the transcriptional start site of the gene encoding the Pol III-specific subunit C53 (CG5147EY22749, henceforth called dC53EY, was backcrossed into a healthy, outbred population of flies. Homozygous dC53EY/EY mutants were not viable, but heterozygous females had reduced dC53 mRNA levels and lived longer than controls. Taken together, these data strongly indicate that Pol III limits lifespan in multiple model organisms and conversely, that partial inhibition of its activity is an intervention that increases longevity in multiple species (Filer, 2017).
The longevity of an animal can be governed from a single organ. In the worm, this role is often played by the gut. To restrict the rpc-1 knockdown to the gut, worms were used that were deficient in rde-1, in which the RNAi machinery deficiency is restored in the gut by gut-specific rde-1 rescue. rpc-1 RNAi extended the lifespan of this strain, both at 20°C and 25°C. Similarly, in the adult fly, driving an RNAi construct targeting the RPC160 orthologue (CG17209, henceforth called dC160,with the mid-gut-specific, RU486-inducible driver TIGS extended the lifespan of females, while the presence of the inducer (RU486) did not affect survival of the control strains. The longevity phenotype could also be recapitulated with RNAi against dC53, another Pol III subunit, indicating that the phenotype was not subunit-specific or due to off-target effects. As well as the gut, longevity can also be associated with the fat body and neurons in flies. However, the longevity phenotype caused by dC160 RNAi appears to be specific to the gut, since no significant lifespan extension was observed upon induction of dC160 RNAi in the fat body of the adult fly, and only a modest, albeit significant, extension resulted from neuronal induction of dC160 RNAi (Filer, 2017).
The worm gut is composed of only post-mitotic cells. In flies, as in mammals, the adult gut epithelium contains mitotically active intestinal stem cells, and the mid-gut-specific driver TIGS appears to be active in at least some ISCs, prompting restricting of dC160 RNAi induction to this cell type. ISC-specific dC160 RNAi, achieved with the GS5961 driver, was sufficient to promote longevity. In summary, Pol III activity in the gut limits survival in worms and flies, and in the fly, Pol III can drive ageing specifically from the gut stem-cell compartment (Filer, 2017).
The consequences of Pol III inhibition in the fly gut was assessed. Pol III acts to generate precursor tRNAs (pre-tRNAs) that are processed rapidly to mature tRNAs. Owing to their short half-lives, pre-tRNAs are useful as readouts of in vivo Pol III activity. Profiling the levels of specific pre-tRNAs, pre-tRNAHis, pre-tRNAAla and pre-tRNALeu, relative to the levels of U3 (a small nucleolar RNA transcribed by Pol II) revealed a moderate but significant reduction in Pol III activity upon gut-specific induction of dC160 RNAi. The three polymerases can be directly coordinated to generate the translation machinery. Indeed, Pol III inhibition had knock-on effects on Pol I- but not Pol II-generated transcripts, revealing partial cross-talk. dC160 RNAi also reduced protein synthesis in the gut, consistent with reduced Pol III activity. These effects (reduction in pre-tRNAs or protein synthesis) were not observed after feeding RU486 to the driver-only control. The reduction in protein synthesis was not pathological: total protein content of the gut was unaltered; fecundity, a sensitive readout of a female's nutritional status, was unaffected; and the flies' weight, triacylglycerol and protein levels remained unchanged. Reduced protein synthesis can liberate protein-folding machinery from protein production and increase homeostatic capacity. Indeed, induction of dC160 RNAi in the gut increased the resistance of adult flies to proteostatic challenge with tunicamycin for TIGS-only control. Hence, Pol III can fine-tune the rate of protein synthesis in the adult fly gut without obvious detrimental outcomes, while increasing resistance to proteotoxic stress (Filer, 2017).
Having demonstrated the relevance of Pol III for ageing, whether it acts on lifespan downstream of TORC1 was investigated in Drosophila. Numerous observations in several organisms support the model in which TORC1 localizes on Pol III-transcribed loci and promotes phosphorylation of the components of the Pol III transcriptional machinery to activate transcription, in part by inhibition of the Pol III repressor, Maf1. Using chromatin immunoprecipitation (ChIP) with two independently generated antibodies against Drosophila TOR (target of rapamycin), TOR enrichment was observed on Pol III-target genes in the adult fly, relative to Pol II targets. Inhibition of TORC1 by feeding rapamycin to flies reduced the levels of pre-tRNAs in whole flies. Rapamycin also reduced pre-tRNA levels specifically in the gut relative to U3. Since rapamycin results in re-scaling of the gut, evidenced by the reduction in the total RNA content of the organ, it was also confirmed that the drug reduced pre-tRNA levels relative to total RNA. Interestingly, rapamycin did not cause a decrease in 45S pre-rRNA in the gut, suggesting a lack of sustained Pol I inhibition. Additionally, gut-specific overexpression of Maf1 reduced the levels of pre-tRNAs and extended lifespan, confirming that Maf1 acts on Pol III in the adult gut. These data are consistent with TORC1 driving systemic and gut-specific Pol III activity in the adult fruitfly (Filer, 2017).
To examine whether the lifespan effects of Pol III are downstream of TORC1, adult-onset Pol III inhibition was combined with rapamycin treatment. Rapamycin feeding or gut-specific dC160 RNAi resulted in the same magnitude of lifespan extension. The two treatments were not additive, consistent with their acting on the same longevity pathway. The same effect was observed with RNAi against dC53 in the gut, as well as when dC160 RNAi was restricted to the ISCs. Importantly, rapamycin feeding also inhibited phosphorylation of the TORC1 substrate, S6 kinase (S6K), in both the gut and the whole fly, and decreased fecundity, while gut-specific C160 RNAi did not have these effects. This confirms that Pol III inhibition does not impact TORC1 activity locally or systemically, and therefore, Pol III acts downstream of TORC1 in ageing (Filer, 2017).
TORC1 inhibition is known to ameliorate age-related pathology and functional decline of the gut. Whether inhibition of Pol III was sufficient to block the dysplasia resulting from hyperproliferation and aberrant differentiation of ISCs was examined by assessing the characteristic, age-dependent increase in dividing phospho-histone H3 (pH3)-positive cells. Inducing dC160 RNAi in the fly gut or solely in the ISCs ameliorated this pathology. These treatments also counteracted the age-related loss of gut barrier function, decreasing the number of flies displaying extra-intestinal accumulation of a blue food dye (the 'Smurf' phenotype). It as also found that rpc-1 RNAi reduced the severity of age-related loss of gut-barrier function in worms. In Drosophila, gut health and TORC1 inhibition are specifically linked to female survival. Indeed, induction of dC160 RNAi in the gut had a sexually dimorphic effect on lifespan, as the effect on males, although significant, was lower in magnitude relative to the effect on females. Overall, the data show that gut- or ISC-specific inhibition of Pol III, which extends lifespan, is sufficient to ameliorate age-related impairments in gut health, which may be causative of or correlate with this longevity (Filer, 2017).
This study demonstrates that the adult-onset decrease in the growth-promoting anabolic function mediated by Pol III in the gut, and specifically in the intestinal stem-cell compartment, is sufficient to recapitulate the longevity benefits of rapamycin treatment. Pol III activity is essential for growth; its detrimental effects on ageing suggest an antagonistic pleiotropy in which wild-type levels of Pol III activity are optimised for growth and reproductive fitness in early life but prove detrimental for later health. This study reveals a fundamental role for Pol III in adult physiology, implicating wild-type Pol III activity in age-related stem-cell dysfunction, declining gut health and organismal survival downstream of nutrient signalling pathways. The longevity resulting from partial Pol III inhibition in adulthood is likely to result from the reduced provision of protein synthetic machinery; however, differential regulation of tRNA genes or Pol III-mediated changes to chromatin organization may also be involved, as has been suggested in other contexts (Arimbasseri, 2016). The strong structural and functional conservation of Pol III in eukaryotes suggests that studies of its influence on mammalian ageing are warranted and could lead to important therapies (Filer, 2017).
The Drosophila ovary is a widely used model for germ cell and somatic tissue biology. This study used single-cell RNA-sequencing (scRNA-seq) to build a comprehensive cell atlas of the adult Drosophila ovary that contains transcriptional profiles for every major cell type in the ovary, including the germline stem cells and their niche cells, follicle stem cells, and previously undescribed subpopulations of escort cells. In addition, Gal4 lines were identified with specific expression patterns, and lineage tracing of subpopulations of escort cells and follicle cells was performed. A distinct subpopulation of escort cells was capable of converting to follicle stem cells in response to starvation or upon genetic manipulation, including knockdown of escargot, or overactivation of mTor or Toll signalling (Rust, 2020).
In summary, this study has generated a detailed atlas of the cells in the adult Drosophila ovary. This atlas consists of 26 clusters that each correspond to a distinct population in the ovary. Through experimental validation and referencing well-characterized markers in the literature, the identity of each cluster was identified, and all of the major cell types in the ovariole were found to be represented. Several transcriptionally distinct subpopulations were identified within these major cell types, such as the anterior, central, and posterior Escort cell (EC) populations. Both the GSCs and the FSCs were identified in the dataset, which revealed several genes that are predicted to be specific for each of these stem cell populations. In addition, several Gal4 drivers, including Pdk1-Gal4, fax-Gal4, and stl-Gal4, were identified with unique expression patterns that make it possible to target transgene expression to the subsets of cells marked by these drivers. Lastly, although this study primarily focused on the most uniquely expressed genes for each cluster in this study, the transcriptional profile of each cluster is a rich dataset that can be mined to identify populations of cells that are relevant for a topic of interest. For example, gene expression profile of each cluster was compared to a list of human disease genes that are well-suited for analysis in Drosophila. It was found that germ cells are enriched for cells expressing major drivers of cancer, and ECs and follicle cells are enriched for genes involved in cardiac dysfunction, suggesting that these cell types may be good starting points for studies into the genetic interactions that underlie these human diseases (Rust, 2020).
This study also demonstrates the utility of using CellFindR9 in combination with monocle320 to identify unique populations of cells within a dataset. Because CellFindR produces clusters in a structured, iterative fashion, it was possible to construct a hierarchical tree that corresponds to a transcriptome relationship between clusters, and this outperformed other clustering methods. The tree built by CellFindR aligns well with expectations and provides some interesting new insights. For example, it was expected that germ cells would cluster apart from somatic cells in Tier 1 because these populations are substantially different from each other, arising at different times during development and from completely different lineages. However, it was surprising that the FSC, pFCs, polar cells, and stalk cells clustered more closely to escort cells than to the follicle cells of budded follicles. This suggests that many cell types in the germarium, which are often studied separately, have biologically relevant similarities (Rust, 2020).
The use of G-TRACE to assess the lineage potential of somatic cells in the germarium led to the surprising finding that ECs can convert to FSCs under starvation conditions. Recent studies have described similar forms of cellular plasticity in other tissues, suggesting that the ability of non-stem cells to convert to stem cells may be a more general feature of adult stem cell niches. However, this aspect of tissue homeostasis remains poorly understood. The finding that the conversion of ECs to FSCs can be induced by perturbations of mTor or Toll signaling is consistent with a role for these pathways in responding to starvation and cellular stress in other tissues, and provides a new opportunity to investigate the mechanisms of cellular responses to physiological stress in an adult stem cell niche (Rust, 2020).
Overall, this study provides a resource that will be valuable for a wide range of studies that use the Drosophila ovary as an experimental model. Additional scRNA-seq datasets provided by other studies will further increase the accuracy and resolution of the ovary cell atlas, and it will be important to follow up on the predictions of the atlas with detailed studies that focus on specific populations of cells. Collectively, these efforts will help drive discovery forward by providing a deeper understanding of the cellular composition of the Drosophila ovary (Rust, 2020).
FK506 and rapamycin are related immunosuppressive compounds that block helper T cell activation by interfering with signal transduction. In vitro, both drugs bind and inhibit the FK506-binding protein (FKBP) proline rotamase. Saccharomyces cerevisiae cells treated with rapamycin irreversibly arrest in the G1 phase of the cell cycle. An FKBP-rapamycin complex is concluded to be the toxic agent because (1) strains that lack FKBP proline rotamase, encoded by FPR1, are viable and fully resistant to rapamycin and (2) FK506 antagonizes rapamycin toxicity in vivo. Mutations that confer rapamycin resistance alter conserved residues in FKBP that are critical for drug binding. Two genes other than FPR1, named TOR1 and TOR2, that participate in rapamycin toxicity have been identified. Nonallelic noncomplementation between FPR1, TOR1, and TOR2 alleles suggests that the products of these genes may interact as subunits of a protein complex. Such a complex may mediate nuclear entry of signals required for progression through the cell cycle (Heitman, 1991).
Saccharomyces cerevisiae cells treated with the immunosuppressant rapamycin or depleted for the targets of rapamycin TOR1 and TOR2 arrest growth in the early G1 phase of the cell cycle. Loss of TOR function also causes an early inhibition of translation initiation and induces several other physiological changes characteristic of starved cells entering stationary phase (G0). A G1 cyclin mRNA whose translational control is altered by substitution of the UBI4 5' leader region (UBI4 is normally translated under starvation conditions) suppresses the rapamycin-induced G1 arrest and confers starvation sensitivity. These results suggest that the block in translation initiation is a direct consequence of loss of TOR function and the cause of the G1 arrest. It is proposed that the TORs, two related phosphatidylinositol kinase homologs, are part of a novel signaling pathway that activates eIF-4E-dependent protein synthesis and, thereby, G1 progression in response to nutrient availability. Such a pathway may constitute a checkpoint that prevents early G1 progression and growth in the absence of nutrients (Barbet, 1996).
The rapamycin-sensitive TOR signalling pathway in Saccharomyces cerevisiae activates a cell-growth program in response to nutrients such as nitrogen and carbon. The TOR1 and TOR2 kinases (TOR) control cytoplasmic protein synthesis and degradation through the conserved TAP42 protein. Upon phosphorylation by TOR, TAP42 binds and possibly inhibits type 2A and type-2A-related phosphatases; however, the mechanism by which TOR controls nuclear events such as global repression of starvation-specific transcription is unknown. TOR prevents transcription of genes expressed upon nitrogen limitation by promoting the association of the GATA transcription factor GLN3 with the cytoplasmic protein URE2. The binding of GLN3 to URE2 requires TOR-dependent phosphorylation of GLN3. Phosphorylation and cytoplasmic retention of GLN3 are also dependent on the TOR effector TAP42, and are antagonized by the type-2A-related phosphatase SIT4. TOR inhibits expression of carbon-source-regulated genes by stimulating the binding of the transcriptional activators MSN2 and MSN4 to the cytoplasmic 14-3-3 protein BMH2. Thus, the TOR signalling pathway broadly controls nutrient metabolism by sequestering several transcription factors in the cytoplasm (Beck, 1999).
Rapamycin inhibits the TOR kinases, which regulate cell proliferation and mRNA translation and are conserved from yeast to human. The TOR kinases also regulate responses to nutrients, including sporulation, autophagy, mating, and ribosome biogenesis. Gene expression in yeast cells exposed to rapamycin has been analyzed using arrays representing the whole yeast genome. TOR inhibition by rapamycin induces expression of nitrogen source utilization genes controlled by the Ure2 repressor and the transcriptional regulator Gln3, and globally represses ribosomal protein expression. gln3 mutations confer rapamycin resistance, whereas ure2 mutations confer rapamycin hypersensitivity, even in cells expressing dominant rapamycin-resistant TOR mutants. Ure2 is a phosphoprotein in vivo that is rapidly dephosphorylated in response to rapamycin or nitrogen limitation. In summary, these results reveal that the TOR cascade plays a prominent role in regulating transcription in response to nutrients in addition to its known roles in regulating translation, ribosome biogenesis, and amino acid permease stability (Cardenas, 1999).
The immunosuppressant rapamycin inhibits Tor1p and Tor2p (target of rapamycin proteins), ultimately resulting in cellular responses characteristic of nutrient deprivation through a mechanism involving translational arrest. The immediate transcriptional response of yeast grown in rich media and treated with rapamycin was measured to investigate the direct effects of Tor proteins on nutrient-sensitive signaling pathways. The results suggest that Tor proteins directly modulate the glucose activation and nitrogen discrimination pathways and the pathways that respond to the diauxic shift (including glycolysis and the citric acid cycle). Tor proteins do not directly modulate the general amino acid control, nitrogen starvation, or sporulation (in diploid cells) pathways. Poor nitrogen quality activates the nitrogen discrimination pathway, which is controlled by the complex of the transcriptional repressor Ure2p and activator Gln3p. Inhibiting Tor proteins with rapamycin increases the electrophoretic mobility of Ure2p. The work presented here illustrates the coordinated use of genome-based and biochemical approaches to delineate a cellular pathway modulated by the protein target of a small molecule (Hardwick, 1999).
The functional diversity and structural heterogeneity of microtubules are largely determined by microtubule-associated proteins (MAPs). Bik1p (bilateral karyogamy defect protein) is one of the MAPs required for microtubulen assembly, stability and function in cell processes such as karyogamy and nuclear migration and positioning in the yeast Saccharomyces cerevisiae. The macrocyclic immunosuppressive antibiotic rapamycin, complexed with its binding protein FKBP12, binds to and inhibits the target of rapamycin protein (TOR) in yeast. TOR physically interacts with Bik1p, the yeast homolog of human CLIP-170/Restin. Inhibition of TOR by rapamycin significantly affects microtubule assembly, elongation and stability. This function of TOR is independent of new protein synthesis. Rapamycin also causes defects in spindle orientation, nuclear movement and positioning, karyogamy and chromosomal stability, defects also found in the bikDelta mutant. These data suggest a role for TOR signaling in regulating microtubule stability and function, possibly through Bik1p (Choi, 2000).
Rapamycin inhibits pseudohyphal filamentous differentiation of S. cerevisiae in response to nitrogen limitation. Overexpression of Tap42, a protein phosphatase regulatory subunit, restores pseudohyphal growth in cells exposed to rapamycin. The tap42-11 mutation compromises pseudohyphal differentiation and renders it resistant to rapamycin. Cells lacking the Tap42-regulated protein phosphatase Sit4 exhibit a pseudohyphal growth defect and are markedly hypersensitive to rapamycin. Mutations in other Tap42-regulated phosphatases have no effect on pseudohyphal differentiation. These findings support a model in which pseudohyphal differentiation is controlled by a nutrient-sensing pathway involving the Tor protein kinases and the Tap42-Sit4 protein phosphatase. Activation of the MAP kinase or cAMP pathways, or mutation of the Sok2 repressor, restore filamentation in rapamycin treated cells, supporting models in which the Tor pathway acts in parallel with these known pathways. Filamentous differentiation of diverse fungi is also blocked by rapamycin, demonstrating that the Tor signaling cascade plays a conserved role in regulating filamentous differentiation in response to nutrients (Cutler, 2001).
The TOR protein is a phosphoinositide kinase-related kinase widely conserved among eukaryotes. Fission yeast tor1 encodes an ortholog of TOR, which is required for sexual development and growth under stressed conditions. gad8, which encodes a Ser/Thr kinase of the AGC family, was isolated as a high-copy suppressor of the sterility of a tor1 mutant. Disruption of gad8 causes phenotypes similar to those of tor1 disruption. Gad8p is less phosphorylated and its kinase activity is undetectable in tor1Delta cells. Three amino acid residues corresponding to conserved phosphorylation sites in the AGC family kinases, namely Thr387 in the activation loop, Ser527 in the turn motif and Ser546 in the hydrophobic motif, are important for the kinase activity of Gad8p. Tor1p is responsible for the phosphorylation of Ser527 and Ser546, whereas Ksg1p, a PDK1-like kinase, appears to phosphorylate Thr387 directly. Altogether, Tor1p, Ksg1p and Gad8p appear to constitute a signaling module for sexual development and growth under stressed conditions in fission yeast, which resembles the mTOR-PDK1-S6K1 system in mammals and may represent a basic signaling module ubiquitous in eukaryotes (Matsuo, 2003).
The target of rapamycin (TOR) protein is a conserved regulator of ribosome biogenesis, an important process for cell growth and proliferation. However, how TOR is involved remains poorly understood. Rapamycin and nutrient starvation, conditions inhibiting TOR, are found to lead to significant nucleolar size reduction in both yeast and mammalian cells. In yeast, this morphological change is accompanied by release of RNA polymerase I (Pol I) from the nucleolus and inhibition of ribosomal DNA (rDNA) transcription. Evidence is presented that TOR regulates association of Rpd3-Sin3 histone deacetylase (HDAC) with rDNA chromatin, leading to site-specific deacetylation of histone H4. Moreover, histone H4 hypoacetylation mutations cause nucleolar size reduction and Pol I delocalization, while rpd3Delta and histone H4 hyperacetylation mutations block the nucleolar changes as a result of TOR inhibition. Taken together, these results suggest a chromatin-mediated mechanism by which TOR modulates nucleolar structure, RNA Pol I localization and rRNA gene expression in response to nutrient availability (Tsang, 2003).
In complex with FKBP12, the immunosuppressant rapamycin binds to and inhibits the yeast TOR1 and TOR2 proteins and the mammalian homologue mTOR/FRAP/RAFT1. The TOR proteins promote cell cycle progression in yeast and human cells by regulating translation and polarization of the actin cytoskeleton. A C-terminal domain of the TOR proteins shares identity with protein and lipid kinases, but only one substrate (PHAS-I), and no regulators of the TOR-signaling cascade have been identified. Yeast TOR1 has been shown to have an intrinsic protein kinase activity capable of phosphorylating PHAS-1, and this activity is abolished by an active site mutation and inhibited by FKBP12-rapamycin or wortmannin. An intact TOR1 kinase domain is essential for TOR1 functions in yeast. Overexpression of a TOR1 kinase-inactive mutant, or of a central region of the TOR proteins distinct from the FRB and kinase domains, is toxic in yeast, and overexpression of wild-type TOR1 suppresses this toxic effect. Expression of the TOR-toxic domain leads to a G1 cell cycle arrest, consistent with an inhibition of TOR function in translation. Overexpression of the PLC1 gene, which encodes the yeast phospholipase C homologue, suppresses growth inhibition by the TOR-toxic domains. In conclusion, these findings identify a toxic effector domain of the TOR proteins that may interact with substrates or regulators of the TOR kinase cascade and that shares sequence identity with other PIK family members, including ATR, Rad3, Mei-41, and ATM (Alarcon, 1999).
The regulation of ribosome biogenesis in response to environmental conditions is a key aspect of cell growth control. Ribosomal protein (RP) genes are regulated by the nutrient-sensitive, conserved target of rapamycin (TOR) signaling pathway. TOR controls the subcellular localization of protein kinase A (PKA) and the PKA-regulated kinase YAK1. However, the target transcription factor(s) of the TOR-PKA pathway are unknown. Regulation of RP gene transcription via TOR and PKA in yeast involves the Forkhead-like transcription factor FHL1 and the two cofactors IFH1 (a coactivator) and CRF1 (a corepressor). TOR, via PKA, negatively regulates YAK1 and maintains CRF1 in the cytoplasm. Upon TOR inactivation, activated YAK1 phosphorylates and activates CRF1. Phosphorylated CRF1 accumulates in the nucleus and competes with IFH1 for binding to FHL1 at RP gene promoters, and thereby inhibits transcription of RP genes. Thus, a signaling mechanism is described linking an environmental sensor to ribosome biogenesis (Martin, 2004).
Calorie restriction increases life span in many organisms, including the budding yeast Saccharomyces cerevisiae. From a large-scale analysis of 564 single-gene-deletion strains of yeast, 10 gene deletions were identified that increase replicative life span. Six of these correspond to genes encoding components of the nutrient-responsive TOR and Sch9 pathways. Calorie restriction of tor1D or sch9D cells failed to further increase life span and, like calorie restriction, deletion of either SCH9 or TOR1 increased life span independent of the Sir2 histone deacetylase. It is proposed that the TOR and Sch9 kinases define a primary conduit through which excess nutrient intake limits longevity in yeast (Kaeberlein, 2005).
Chronological life span (CLS) in Saccharomyces cerevisiae, defined as the time cells in a stationary phase culture remain viable, has been proposed as a model for the aging of post-mitotic tissues in mammals. A high-throughput assay was developed to determine CLS for approximately 4800 single-gene deletion strains of yeast, and long-lived strains were identified carrying mutations in the conserved TOR pathway. TOR signaling regulates multiple cellular processes in response to nutrients, especially amino acids, raising the possibility that decreased TOR signaling mediates life span extension by calorie restriction. In support of this possibility, removal of either asparagine or glutamate from the media significantly increased stationary phase survival. Pharmacological inhibition of TOR signaling by methionine sulfoximine or rapamycin also increased CLS. Decreased TOR activity also promoted increased accumulation of storage carbohydrates and enhanced stress resistance and nuclear relocalization of the stress-related transcription factor Msn2. It is proposed that up-regulation of a highly conserved response to starvation-induced stress is important for life span extension by decreased TOR signaling in yeast and higher eukaryotes (Powers, 2005).
The highly conserved target-of-rapamycin (TOR) protein kinases control cell growth in response to nutrients and growth factors. In mammals, TOR has been shown to interact with raptor (regulatory associated protein of mTOR: potential Drosophila homolog CG4320) to relay nutrient signals to downstream translation machinery. Raptor associates in a near stoichiometric ratio with mTOR to form a complex that functions as the nutrient sensor. It was proposed that raptor acts as a scaffold to bridge TOR with its putative phosphorylation targets. In C. elegans, mutations in the genes encoding CeTOR and raptor result in dauer-like larval arrest, implying that CeTOR regulates dauer diapause. The daf-15 (raptor) and let-363 (CeTOR) mutants shift metabolism to accumulate fat, and raptor mutations extend adult life span. daf-15 transcription is regulated by DAF-16, a FOXO transcription factor that is in turn regulated by daf-2 insulin/IGF signaling. This is a new mechanism that regulates the TOR pathway. Thus, DAF-2 insulin/IGF signaling and nutrient signaling converge on DAF-15 (raptor) to regulate C. elegans larval development, metabolism and life span (Jia, 2004).
FoxA factors are critical regulators of embryonic development and postembryonic life, but little is know about the upstream pathways that modulate their activity. C. elegans pha-4 encodes a FoxA transcription factor that is required to establish the foregut in embryos and to control growth and longevity after birth. The AAA+ ATPase homolog ruvb-1 has been identified as a potent suppressor of pha-4 mutations. This study shows that ruvb-1 is a component of the Target of Rapamycin (TOR) pathway in C. elegans (CeTOR). Both ruvb-1 and let-363/TOR control nucleolar size and promote localization of box C/D snoRNPs to nucleoli, suggesting a role in rRNA maturation. Inactivation of let-363/TOR or ruvb-1 suppresses the lethality associated with reduced pha-4 activity. The CeTOR pathway controls protein homeostasis and also contributes to adult longevity. This study found that pha-4 is required to extend adult lifespan in response to reduced CeTOR signaling. Mutations in the predicted CeTOR target rsks-1/S6 kinase or in ife-2/eIF4E also reduce protein biosynthesis and extend lifespan, but only rsks-1 mutations require pha-4 for adult longevity. In addition, rsks-1, but not ife-2, can suppress the larval lethality associated with pha-4 loss-of-function mutations. In conclusion this data suggests that pha-4 and the CeTOR pathway antagonize one another to regulate postembryonic development and adult longevity. A model is suggested in which nutrients promote TOR and S6 kinase signaling, which represses pha-4/FoxA, leading to a shorter lifespan. A similar regulatory hierarchy may function in other animals to modulate metabolism, longevity, or disease (Sheaffer, 2008).
Rictor is a component of the target of rapamycin complex 2 (TORC2). While TORC2 has been implicated in insulin and other growth factor signaling pathways, the key inputs and outputs of this kinase complex remain unknown. Mutations have been identified in the C. elegans homolog of rictor in a forward genetic screen for increased body fat. Despite high body fat, rictor mutants are developmentally delayed, small in body size, lay an attenuated brood, and are short-lived, indicating that Rictor plays a critical role in appropriately partitioning calories between long-term energy stores and vital organismal processes. Rictor is also necessary to maintain normal feeding on nutrient-rich food sources. In contrast to wild-type animals, which grow more rapidly on nutrient-rich bacterial strains, rictor mutants display even slower growth, a further reduced body size, decreased energy expenditure, and a dramatically extended life span, apparently through inappropriate, decreased consumption of nutrient-rich food. Rictor acts directly in the intestine to regulate fat mass and whole-animal growth. Further, the high-fat phenotype of rictor mutants is genetically dependent on akt-1, akt-2, and serum and glucocorticoid-induced kinase-1 (sgk-1). Alternatively, the life span, growth, and reproductive phenotypes of rictor mutants are mediated predominantly by sgk-1. These data indicate that Rictor/TORC2 is a nutrient-sensitive complex with outputs to AKT and SGK to modulate the assessment of food quality and signal to fat metabolism, growth, feeding behavior, reproduction, and life span (Soukas, 2009).
The evolutionarily conserved target of rapamycin (TOR) kinase controls fundamental metabolic processes to support cell and tissue growth. TOR functions within the context of two distinct complexes, TORC1 and TORC2. TORC2, with its specific component Rictor, has been recently implicated in aging and regulation of growth and metabolism. This study identified rict-1/Rictor (homolog of Drosophila Rictor) as a regulator of embryonic development in C. elegans. The transcription factor skn-1 establishes development of the mesendoderm in embryos, and is required for cellular homeostasis and longevity in adults. Loss of maternal skn-1 function leads to mis-specification of the mesendodermal precursor and failure to form intestine and pharynx. Genetic inactivation of rict-1 suppressed skn-1-associated lethality by restoring mesendodermal specification in skn-1 deficient embryos. Inactivation of other TORC2 but not TORC1 components also partially rescues skn-1 embryonic lethality. The SGK-1 kinase (homolog of the vertebate serum- and glucocorticoid-inducible kinase SGK) mediates these functions downstream of rict-1/TORC2, as a sgk-1 gain-of-function mutant suppresses the rict-1 mutant phenotype. These data indicate that TORC2 and SGK-1 antagonize SKN-1 during embryonic development (Ruf, 2013).
Metazoans adapt to a low-oxygen environment (hypoxia) through activation of stress-response pathways. This study reports that transient hypoxia exposure extends lifespan in C. elegans through mitochondrial reactive oxygen species (ROS)-dependent regulation of the nutrient-sensing kinase target of rapamycin (TOR) and its upstream activator, RHEB-1 (see Drosophila Rheb). The increase in lifespan during hypoxia requires the intestinal GATA-type transcription factor ELT-2 (see Drosophila GATAe) downstream of TOR signaling. Using RNA sequencing (RNA-seq), this study describes an ELT-2-dependent hypoxia response that includes an intestinal glutathione S-transferase, GSTO-1, and it was uncovered that GSTO-1 is required for lifespan under hypoxia. These results indicate mitochondrial ROS-dependent TOR signaling integrates metabolic adaptations in order to confer survival under hypoxia (Schieber, 2014). .
The structurally related natural products rapamycin and FK506 bind to the same intracellular receptor, FKBP12, yet the resulting complexes interfere with distinct signalling pathways. FKBP12-rapamycin inhibits progression through the G1 phase of the cell cycle in osteosarcoma, liver and T cells as well as in yeast, and interferes with mitogenic signalling pathways that are involved in G1 progression, namely with activation of the protein p70S6k and cyclin-dependent kinases. A mammalian FKBP-rapamycin-associated protein (FRAP) has been isolated whose binding to structural variants of rapamycin complexed to FKBP12 correlates with the ability of these ligands to inhibit cell-cycle progression. Peptide sequences from purified bovine FRAP were used to isolate a human cDNA clone that is highly related to the DRR1/TOR1 and DRR2/TOR2 gene products from Saccharomyces cerevisiae. Although it has not been previously demonstrated that either of the DRR/TOR gene products can bind the FKBP-rapamycin complex directly, these yeast genes have been genetically linked to a rapamycin-sensitive pathway and are thought to encode lipid kinases (Brown, 1994).
The immunosuppressants rapamycin and FK506 bind to the same intracellular protein, the immunophilin FKBP12. The FKB12-FK506 complex interacts with and inhibits the Ca(2+)-activated protein phosphatase calcineurin. A protein complex containing 245 kDa and 35 kDa components, designated rapamycin and FKBP12 targets 1 and 2 (RAFT1 and RAFT2), interacts with FKBP12 in a rapamycin-dependent manner. Sequences (330 amino acids total) of tryptic peptides derived from the 245 kDa RAFT1 reveal striking homologies to the yeast TOR gene products, which were originally identified by mutations that confer rapamycin resistance in yeast. A RAFT1 cDNA was obtained and found to encode a 289 kDa protein (2549 amino acids) that is 43% and 39% identical to TOR2 and TOR1, respectively. It is proposed that RAFT1 is the direct target of FKBP12-rapamycin and a mammalian homolog of the TOR proteins (Sabatini, 1994).
The effects of insulin on the mammalian target of rapamycin, mTOR, were investigated in 3T3-L1 adipocytes. mTOR protein kinase activity was measured in immune complex assays with recombinant PHAS-I as substrate. Insulin-stimulated kinase activity is clearly observed when immunoprecipitations are conducted with the mTOR antibody, mTAb2. Insulin also increases by severalfold the 32P content of mTOR, determined after purifying the protein from 32P-labeled adipocytes with rapamycin.FKBP12 agarose beads. Insulin affects neither the amount of mTOR immunoprecipitated nor the amount of mTOR detected by immunoblotting with mTAb2. However, the hormone markedly decreases the reactivity of mTOR with mTAb1, an antibody that activates the mTOR protein kinase. The effects of insulin on increasing mTOR protein kinase activity and on decreasing mTAb1 reactivity are abolished by incubating mTOR with protein phosphatase 1. Interestingly, the epitope for mTAb1 is located near the COOH terminus of mTOR in a 20-amino acid region that includes consensus sites for phosphorylation by protein kinase B (PKB). Experiments were performed in MER-Akt cells to investigate the role of PKB in controlling mTOR. These cells express a PKB-mutant estrogen receptor fusion protein that is activated when the cells are exposed to 4-hydroxytamoxifen. Activating PKB with 4-hydroxytamoxifen mimics insulin by decreasing mTOR reactivity with mTAb1 and by increasing the PHAS-I kinase activity of mTOR. These findings support the conclusion that insulin activates mTOR by promoting phosphorylation of the protein via a signaling pathway that contains PKB (Scott, 1999).
Hormones and growth factors induce protein translation in part by phosphorylation of the eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1; see Drosophila Thor). The rapamycin and FK506-binding protein (FKBP)-target 1 (RAFT1, also known as FRAP) is a mammalian homolog of the Saccharomyces cerevisiae target of rapamycin proteins (mTOR) that regulates 4E-BP1. However, the molecular mechanisms involved in growth factor-initiated phosphorylation of 4E-BP1 are not well understood. Protein kinase Cdelta (PKCdelta) associates with RAFT1 and PKCdelta is required for the phosphorylation and inactivation of 4E-BP1. PKCdelta-mediated phosphorylation of 4E-BP1 is wortmannin resistant but rapamycin sensitive. As shown for serum, phosphorylation of 4E-BP1 by PKCdelta inhibits the interaction between 4E-BP1 and eIF4E and stimulates cap-dependent translation. Moreover, a dominant-negative mutant of PKCdelta inhibits serum-induced phosphorylation of 4E-BP1. These findings demonstrate that PKCdelta associates with RAFT1 and thereby regulates phosphorylation of 4E-BP1 and cap-dependent initiation of protein translation (Kumar, 2000a).
The c-Abl protein-tyrosine kinase is activated by ionizing radiation and certain other DNA-damaging agents. The rapamycin and FKBP-target 1 (RAFT1), also known as FKBP12-rapamycin-associated protein (FRAP, mTOR), regulates the p70S6 kinase [p70(S6k)] and the eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1). The present results demonstrate that c-Abl binds directly to RAFT1 and phosphorylates RAFT1 in vitro and in vivo. c-Abl inhibits autophosphorylation of RAFT1 and RAFT1-mediated phosphorylation p70(S6k). The functional significance of the c-Abl-RAFT1 interaction is further supported by the finding that eIF4E-dependent translation in mouse embryo fibroblasts from Abl(-/-) mice is significantly higher than that compared in wild-type cells. The results also demonstrate that exposure of cells to ionizing radiation is associated with c-Abl-mediated binding of 4E-BP1 to eIF4E and inhibition of translation. These findings with the c-Abl tyrosine kinase represent the first demonstration of a negative physiologic regulator of RAFT1-mediated 5' cap-dependent translation (Kumar, 2000b).
Target of Rapamycin (TOR) mediates a signalling pathway that couples amino acid availability to S6 kinase (S6K) activation, translational initiation and cell growth. Tuberous sclerosis 1 (Tsc1) and Tsc2, tumor suppressors that are responsible for the tuberous sclerosis syndrome, antagonize this amino acid-TOR signalling pathway. Tsc1 and Tsc2 can physically associate with TOR and function upstream of TOR genetically. In Drosophila melanogaster and mammalian cells, loss of Tsc1 and Tsc2 results in a TOR-dependent increase of S6K activity. Furthermore, although S6K is normally inactivated in animal cells in response to amino acid starvation, loss of Tsc1-Tsc2 renders cells resistant to amino acid starvation. It is proposed that the Tsc1-Tsc2 complex antagonizes the TOR-mediated response to amino acid availability. These studies identify Tsc1 and Tsc2 as regulators of the amino acid-TOR pathway and provide a new paradigm for how proteins involved in nutrient sensing function as tumor suppressors (Gao, 2002).
TSC1-TSC2 inhibits the p70 ribosomal protein S6 kinase 1 (an activator of translation) and activates the eukaryotic initiation factor 4E binding protein 1 (4E-BP1, an inhibitor of translational initiation). These functions of TSC1-TSC2 are mediated by inhibition of the mammalian target of rapamycin (mTOR). Furthermore, TSC2 is directly phosphorylated by Akt, which is involved in stimulating cell growth and is activated by growth stimulating signals, such as insulin. TSC2 is inactivated by Akt-dependent phosphorylation, which destabilizes TSC2 and disrupts its interaction with TSC1. These data indicate a molecular mechanism for TSC2 in insulin signalling, tumor suppressor functions and in the inhibition of cell growth (Inoki, 2002).
The small G protein Rheb (Ras homolog enriched in brain) is a molecular target of TSC1/TSC2 that regulates mTOR signaling. Overexpression of Rheb activates 40S ribosomal protein S6 kinase 1 (S6K1) but not p90 ribosomal S6 kinase 1 (RSK1) or Akt. Furthermore, Rheb induces phosphorylation of eukaryotic initiation factor 4E binding protein 1 (4E-BP1) and causes 4E-BP1 to dissociate from eIF4E. This dissociation is completely sensitive to rapamycin (an mTOR inhibitor) but not wortmannin (a phosphoinositide 3-kinase [PI3K] inhibitor). Rheb also activates S6K1 during amino acid insufficiency via a rapamycin-sensitive mechanism, suggesting that Rheb participates in nutrient signaling through mTOR. Moreover, Rheb does not activate a S6K1 mutant that is unresponsive to mTOR-mediated signals, confirming that Rheb functions upstream of mTOR. Overexpression of the Tuberin-Hamartin heterodimer inhibits Rheb-mediated S6K1 activation, suggesting that Tuberin functions as a Rheb GTPase activating protein (GAP). Supporting this notion, TSC patient-derived Tuberin GAP domain mutants were unable to inactivate Rheb in vivo. Moreover, in vitro studies reveal that Tuberin, when associated with Hamartin, acts as a Rheb GTPase-activating protein. Finally, it is shown that membrane localization of Rheb is important for its biological activity because a farnesylation-defective mutant of Rheb stimulated S6K1 activation less efficiently. It is concluded that Rheb acts as a novel mediator of the nutrient signaling input to mTOR and is the molecular target of TSC1 and TSC2 within mammalian cells (Tee, 2003).
To examine whether Rheb overexpression could modulate S6K1 activity, S6K1 was coexpressed with Rheb at two different expression levels in HEK293E cells and kinase activity was assayed by using GST-S6 as a substrate. Coexpression of Rheb significantly increased the basal and insulin-stimulated activity of S6K1. Higher levels of Rheb expression enhanced the basal and insulin-stimulated activity of S6K1 by 5.6- and 1.7-fold, respectively, and this activity level is more potent than the S6K1 activity observed in the presence of lower levels of Rheb protein. These results indicate that Rheb activates signaling cascades that result in S6K1 activation (Tee, 2003).
Given that the activity of S6K1 is enhanced upon cell signaling through mTOR, PI3K, and mitogen-activated protein kinase (MAPK) and protein kinase C (PKC)-mediated pathways, it was determined which signaling pathway was activated upon Rheb overexpression. To investigate PI3K-mediated signaling, Rheb, along with Akt, a downstream target of PI3K, was expressed within HEK293E cells and kinase activity was assayed. Whereas EGF stimulation led to a 4-fold increase in Akt activity, Rheb overexpression did not enhance basal or EGF-stimulated Akt activity. In contrast, Rheb potently activated S6K1 by 11-fold when assayed in parallel. Wortmannin was used as a control to show that PI3K-mediated activation of Akt upon stimulation with EGF was specifically measured. To examine whether Rheb enhanced MAPK-mediated signaling, Rheb was coexpressed with RSK1, a known downstream signaling component of MAPK, within HEK293E cells. Although EGF stimulation led to a 12-fold increase in RSK1 activity, Rheb did not augment the basal or EGF-induced activation of RSK1 assayed with GST-S6 as a substrate. In contrast, Rheb overexpression drastically increased S6K1 activity when assayed in parallel. To inhibit activation of ERK (extracellular signal-regulated kinase) through MEK (MAPK/ERK-kinase)-mediated signaling, cells were treated with the U0126 compound to specifically inhibit MEK. These findings suggest that Rheb does not function upstream of either PI3K/Akt or ERK/RSK1 signaling pathways (Tee, 2003).
Because Rheb enhances the activity of S6K1, a downstream component of mTOR, the effects were investigated of Rheb-mediated signaling on 4E-BP1, another downstream component of mTOR. Dephosphorylated species of 4E-BP1 bind to and inhibit eIF4E-driven cap-dependent translation. Phosphorylation of 4E-BP1 at multiple Ser/Thr-Pro residues upon mitogenic stimulation led to the release of 4E-BP1 from eIF4E, which is blocked by both rapamycin and nutrient starvation. Three different phosphorylated species of 4E-BP1 resolve on SDS-PAGE, with gamma- and alpha-isoforms being the most and least phosphorylated species, respectively. To determine whether Rheb activates mTOR- or PI3K-mediated signaling, Rheb was coexpressed with hemagglutinin (HA)-tagged 4E-BP1 in the presence of either rapamycin or wortmannin to inhibit mTOR or PI3K, respectively. Insulin-induced Akt phosphorylation on Ser473 was blocked by wortmannin, revealing that the concentration of wortmannin used in this study efficiently inhibits PI3K-mediated signaling. Insulin-induced phosphorylation of 4E-BP1 was also blocked by wortmannin, as observed by the reduced mobility shift of 4E-BP1 to the less-phosphorylated isoforms and by decreased Ser65 phosphorylation. Rheb overexpression within serum-starved cells potently enhances 4E-BP1 phosphorylation, which was still sensitive to rapamycin. In contrast, treatment of cells with wortmannin was modestly effective at reducing 4E-BP1 phosphorylation upon Rheb overexpression, indicating that PI3K signaling is not essential for Rheb-induced 4E-BP1 phosphorylation. These data suggest that Rheb signals by using an mTOR-dependent rather than a PI3K-dependent mechanism. Rheb-induced 4E-BP1 phosphorylation should promote the release of 4E-BP1 from eIF4E. To confirm this, endogenous eIF4E was purified on m7GTP-Sepharose, which mimics the cap-structure found at the extreme 5' terminus of most cytoplasmic mRNAs, and how much HA-tagged 4E-BP1 was bound to eIF4E was examined. As expected, 4E-BP1 is released from eIF4E upon Rheb overexpression, implying that Rheb activates cap-dependent translation (Tee, 2003).
The above data imply that Rheb may modulate mTOR signaling. Given that the loss of Rheb in yeast mimics nutrient starvation, Rheb overexpression may promote mTOR signaling through a nutrient-regulated signaling pathway. To address this possibility, whether Rheb promotes S6K1 activation was investigated in the absence of amino acids. During conditions of amino acid withdrawal, Rheb overexpression potently activated S6K1, which was completely blocked by rapamycin but only partially inhibited by wortmannin. Importantly, insulin stimulation of these amino acid-deprived cells potently activated Akt (as observed by Akt phosphorylation on Ser473) but only weakly activated S6K1. Therefore, unlike Rheb-mediated signaling, acute stimulation of PI3K and Akt is not sufficient to fully activate S6K1 during nutrient insufficiency. The modest insulin-induced activation of S6K1 during amino acid insufficiency was blocked by wortmannin, revealing that this activation is completely dependent on PI3K. Activation of S6K1 upon readdition of amino acids was enhanced when Rheb was overexpressed or when cells were stimulated with insulin). Interestingly, Rheb-induced S6K1 activation upon readdition of amino acids was completely inhibited by rapamycin but only partially inhibited by wortmannin. In contrast, insulin-induced S6K1 activity was markedly impaired by both rapamycin and wortmannin. These data convincingly reveal that Rheb potently activates S6K in the absence of nutrients through mTOR rather than PI3K-mediated signaling. Therefore, it is likely that Rheb enhances nutrient-mediated signaling through mTOR (Tee, 2003).
To decisively determine whether Rheb positively activates mTOR signaling, use was made of a rapamycin-resistant mutant of S6K1 (S6K1-F5A-DeltaCT). Unlike treatment with wortmannin, rapamycin treatment was unable to prevent insulin-induced activation of S6K1-F5A-DeltaCT, demonstrating that this mutant is responsive to PI3K signaling but not mTOR signaling. As a positive control, PDK1 and PKCζ, which are known to activate S6K1 through a PI3K-dependent input, were coexpressed. Overexpression of Rheb potently activates wild-type S6K1 basally (by 11-fold) and during insulin stimulation (by 17-fold) but does not enhance the activity of the S6K1-F5A-DeltaCT mutant. In contrast, increased PI3K-mediated signaling toward S6K1 by coexpression of PDK1 and PKCζ results in significantly enhanced activation of both wild-type S6K1 and S6K1-F5A-DeltaCT. These findings strongly suggest that Rheb induces S6K1 activation via a signaling input that is upstream of mTOR but not PI3K (Tee, 2003).
Overexpression of wild-type Rheb has been shown to lead to a significant increase in its activity and implies that the majority of the overexpressed Rheb must exist in the active GTP bound form. If this is true, then the RhebGAP activity must be a limiting factor. If Tuberin possesses RhebGAP activity, overexpression of Tuberin should switch Rheb from an active GTP bound state to an inactive GDP bound state. To indirectly measure Rheb activity, Rheb-induced S6K1 activation was analyzed within nutrient-deprived HEK293E cells. Coexpression of Hamartin and Tuberin completely block Rheb's ability to activate S6K1, implying that Tuberin may function as a RhebGAP (Tee, 2003).
If the GAP domain of Tuberin is essential for Rheb inactivation, then patient-derived TSC2 GAP domain point mutants should not block Rheb-induced S6K1 activation. To address this, three Tuberin mutants were generated that mimic patient-derived TSC2 mutations that occur within the GAP domain and these Tuberin mutants were coexpressed with Hamartin and S6K1. Under serum-starved conditions, Rheb potently activates S6K1, which was fully blocked by coexpression of wild-type Tuberin with Hamartin. In contrast, the three TSC2 GAP domain point mutants were unable to repress Rheb-induced S6K1 activation, revealing that the GAP domain of Tuberin is critical for Tuberin's ability to repress Rheb-mediated signaling (Tee, 2003).
The in vivo overexpression data strongly suggest that the Tuberin-Hamartin heterodimer inhibits Rheb function. In order to test whether this is a direct inhibition due to the GAP activity of Tuberin, in vitro GAP assays were performed on purified Rheb. Flag-tagged Hamartin and Flag-tagged Tuberin were expressed separately or together in HEK293 cells and the respective protein(s) were immunoprecipitated for use in Rheb-GAP assays. Interestingly, immunoprecipitated Hamartin or Tuberin display nearly identical GAP activity toward Rheb; both enhance the intrinsic GTPase activity of Rheb by approximately 2-fold. This suggests that a complex between Tuberin and Hamartin is essential for Tuberin's GAP activity toward Rheb and that endogenous levels of coimmunoprecipitating Tuberin or Hamartin are limiting in these reactions. In support of this, coexpression and immunoprecipitation of both Tuberin and Hamartin result in immune complexes that enhance Rheb GTPase activity by more than 100-fold over the activity of either alone and approximately 200-fold over intrinsic Rheb activity. This dramatic increase in GAP activity is detected despite no significant difference in the amount of Tuberin immunoprecipitated when expressed alone or with Hamartin. Therefore, Tuberin and Hamartin together form a GTPase-activating protein complex that greatly enhances the intrinsic GTPase activity of Rheb (Tee, 2003).
In order to test if this activity is potentially important in the prevention of the TSC disease, the GAP activity of wild-type Tuberin was compared to that of a patient-derived mutant mapped to the Tuberin GAP domain (N1651S). Compared to wild-type Tuberin, the Tuberin(N1651S) mutant is greatly reduced in its ability to enhance Rheb GTPase activity. This suggests that there is a correlation in the ability of Tuberin to act as a GAP toward Rheb and its ability to suppress the TSC disease (Tee, 2003).
Farnesylation of Rheb is required for cell cycle progression of S. pombe. To investigate whether farnesylation is important for Rheb's ability to activate S6K1, a farnesylation-defective Rheb(C182S) mutant was generated, in which the cysteine within the farnesylation CAAX motif is substituted for a serine. When overexpressed, the Rheb(C182S) mutant is less efficient at enhancing S6K1 activity than wild-type Rheb. The mutant Rheb(C182S) protein migrated as the upper band on SDS-PAGE, which indicates that it is not being prenylated and is consistent with earlier studies showing that prenylated Rheb migrates more quickly on SDS-PAGE. In contrast, the majority of wild-type Rheb resolved as the lower prenylated band. These findings suggest that the membrane localization of Rheb through farnesylation is important for Rheb to efficiently augment mTOR-mediated signaling (Tee, 2003).
Thus, Rheb functions upstream of mTOR within the nutrient signaling pathway. Rheb specifically activates mTOR-mediated signaling rather than cell signaling through MEK/ERK and PI3K, as shown by Rheb-mediated activation of S6K1 but not Akt or RSK1. Therefore, it is unlikely that Rheb activates PI3K and Raf, two downstream effectors of Ras. Rheb has previously been shown to interact with Raf in vitro, but the current data suggest that Raf is not an effector of Rheb in vivo. Additionally, Rheb overexpression does not increase the activity of the rapamycin-resistant S6K1 mutant that is unresponsive to mTOR signaling inputs but is activated in response to PI3K signaling. S6K1 activation is regulated by multiple signaling inputs, one of which is directed by PI3K. Therefore, these findings are important and confirm that Rheb overexpression specifically promotes mTOR rather than PI3K signaling. Furthermore, Rheb-induced 4E-BP1 phosphorylation is completely sensitive to rapamycin but not to wortmannin, which further strengthens the notion that Rheb acts upstream of mTOR rather than PI3K. 4E-BP1 dissociates from eIF4E upon Rheb overexpression, revealing that Rheb-mediated signaling through mTOR promotes cap-dependent translation (Tee, 2003).
Evidence has also been provided that Rheb functions within the nutrient signaling cascade upstream of mTOR, as shown by Rheb's ability to potently stimulate S6K1 activity during amino acid insufficiency. During amino acid withdrawal, acute insulin stimulation is still able to elicit high levels of Akt phosphorylation but poorly activates S6K1, showing that the nutrient-mediated mTOR signaling input is essential for optimal S6K1 activation. Therefore, Rheb overexpression supersedes the dependency of the nutrient input to mTOR, suggesting that Rheb is an activator of mTOR within the nutrient-signaling pathway. Interestingly, resupplying cells with amino acids further enhances the activity of S6K1 when Rheb is overexpressed, suggesting that amino acids may promote the activation of Rheb. This research has revealed that the Tuberin-Hamartin heterodimer functions as an inhibitor of nutrient signaling through mTOR. The Tuberin-Hamartin heterodimer inhibits Rheb-induced S6K1 activation during conditions of amino acid withdrawal. This work, therefore, extends these earlier studies, revealing that inhibition of Rheb is the mechanism by which the Tuberin-Hamartin heterodimer inhibits nutrient-mediated signaling. Importantly, the Rheb-inhibitory function of Tuberin-Hamartin heterodimers depends on an intact Tuberin GAP domain; patient-derived point mutations within the GAP domain of TSC2 prevented the Tuberin-Hamartin heterodimer from blocking Rheb-induced S6K1 activation. These data indicate that the GAP activity of Tuberin promotes inactivation of Rheb in vivo, presumably through increasing the intrinsic GTPase activity of Rheb. Confirming this hypothesis, in vitro Rheb GTPase activity assays have revealed that Tuberin enhances the intrinsic GTPase activity of Rheb. Interestingly, coexpression of Hamartin with Tuberin markedly enhances the GTPase activity of Rheb, implying that Hamartin promotes the GAP function of Tuberin toward Rheb. A model is proposed whereby Rheb promotes mTOR signaling when it is in an active GTP bound form, whereas the Tuberin-Hamartin heterodimer inhibits Rheb by converting it to an inactive GDP bound state. These findings reveal that the Tuberin-Hamartin heterodimer and Rheb respectively inhibit and activate the nutrient-signaling input to mTOR. Small G proteins are additionally regulated by guanine nucleotide exchange factors (GEFs). In this model it is proposed that a RhebGEF becomes activated during conditions of nutrient sufficiency, and its activation switches Rheb to an active GTP bound form. Therefore, identifying this Rheb-GEF will be of great importance and may provide new insights into how mTOR senses intracellular amino acids. However, at this point the possibility that Rheb- and nutrient-mediated signaling may function in parallel pathways upstream of mTOR cannot be ruled out. Further experiments will be carried out to investigate this possibility (Tee, 2003).
Inorganic polyphosphate (poly P: chains of hundreds of phosphate residues linked by 'high-energy' bonds as in ATP) has been conserved from prebiotic times in all cells. Poly P is essential for a wide variety of functions in bacteria, including virulence in pathogens. In this study, the unique and many-fold stimulation by poly P in vitro is observed of the protein kinase mTOR (mammalian target of rapamycin). To explore the role of poly P in mammalian cells, a yeast polyphosphatase, PPX1, was inserted into the chromosomes of MCF-7 mammary cancer cells. The transfected cells are markedly deficient in their response to mitogens, such as insulin and amino acids, as seen in their failure to activate mTOR to phosphorylate one of its substrates, PHAS-I (the initiation factor 4E-binding protein). In addition, the transfected cells are severely reduced in their growth in a serum-free medium. On the basis of these findings, it is suggested that poly P (and/or PPX1) serves as a regulatory factor in the activation of mTOR in the proliferative signaling pathways of animal cells (Wang, 2003).
Mammalian target of rapamycin (mTOR) is a central regulator of protein synthesis whose activity is modulated by a variety of signals. Energy depletion and hypoxia result in mTOR inhibition. While energy depletion inhibits mTOR through a process involving the activation of AMP-activated protein kinase (AMPK) by LKB1 and subsequent phosphorylation of TSC2, the mechanism of mTOR inhibition by hypoxia is not known. This study shows that mTOR inhibition by hypoxia requires the TSC1/TSC2 tumor suppressor complex and the hypoxia-inducible gene REDD1/RTP801 (Drosophila homologs: scylla and charybde). Disruption of the TSC1/TSC2 complex through loss of TSC1 or TSC2 blocks the effects of hypoxia on mTOR, as measured by changes in the mTOR targets S6K and 4E-BP1, and results in abnormal accumulation of Hypoxia-inducible factor (HIF). In contrast to energy depletion, mTOR inhibition by hypoxia does not require AMPK or LKB1. Down-regulation of mTOR activity by hypoxia requires de novo mRNA synthesis and correlates with increased expression of the hypoxia-inducible REDD1 gene. Disruption of REDD1 abrogates the hypoxia-induced inhibition of mTOR, and REDD1 overexpression is sufficient to down-regulate S6K phosphorylation in a TSC1/TSC2-dependent manner. Inhibition of mTOR function by hypoxia is likely to be important for tumor suppression as TSC2-deficient cells maintain abnormally high levels of cell proliferation under hypoxia (Brugarolas, 2004).
The tuberous sclerosis tumor suppressors TSC1 and TSC2 regulate the mTOR pathway to control translation and cell growth in response to nutrient and growth factor stimuli. The stress response REDD1 gene has been identified as a mediator of tuberous sclerosis complex (TSC)-dependent mTOR regulation by hypoxia. REDD1 inhibits mTOR function to control cell growth in response to energy stress. Endogenous REDD1 is induced following energy stress, and REDD1-/- cells are highly defective in dephosphorylation of the key mTOR substrates S6K and 4E-BP1 following either ATP depletion or direct activation of the AMP-activated protein kinase (AMPK). REDD1 likely acts on the TSC1/2 complex, because regulation of mTOR substrate phosphorylation by REDD1 requires TSC2 and is blocked by overexpression of the TSC1/2 downstream target Rheb but is not blocked by inhibition of AMPK. Tetracycline-inducible expression of REDD1 triggers rapid dephosphorylation of S6K and 4E-BP1 and significantly decreases cellular size. Conversely, inhibition of endogenous REDD1 by short interfering RNA increases cell size in a rapamycin-sensitive manner, and REDD1-/- cells are defective in cell growth regulation following ATP depletion. These results define REDD1 as a critical transducer of the cellular response to energy depletion through the TSC-mTOR pathway (Sofer, 2005).
The mTOR (mammalian target of rapamycin) signalling pathway is a key regulator of cell growth and is controlled by growth factors and nutrients such as amino acids. Although signalling pathways from growth factor receptors to mTOR have been elucidated, the pathways mediating signalling by nutrients are poorly characterized. Through a screen for protein kinases active in the mTOR signalling pathway in Drosophila a Ste20 family member (MAP4K3; Drosophila homolog happyhour) was identified that is required for maximal S6K (S6 kinase)/4E-BP1 [eIF4E (eukaryotic initiation factor 4E)-binding protein 1] phosphorylation and regulates cell growth. Importantly, MAP4K3 activity is regulated by amino acids, but not the growth factor insulin and is not regulated by the mTORC1 inhibitor rapamycin. These results therefore suggest a model whereby nutrients signal to mTORC1 via activation of MAP4K3 (Findley, 2007).
In metazoans both nutrient amino acids and mitogens are required to promote mTOR signalling and cell growth. Activation of mTOR signalling by mitogens involves activation of protein kinases such as PKB, p90RSK and ERK that have been reported to inhibit the function of the TSC1-2 complex which is then thought to lead to increased GTP loading of the Ras-like GTPase Rheb. In contrast, amino acids do not appear to regulate Rheb GTP loading, and, although a class III PI3K has been implicated upstream of mTORC1, no analogous protein kinases have been identified that respond to amino acids and activate mTOR signalling. This study found, both in Drosophila and in mammalian cells, that suppression of MAP4K3 inhibited mTOR signalling to S6K in the context of activation of the pathway induced by deficiency in TSC1-2. Since a variety of oncogenic signalling pathways inhibit TSC1-2, and TSC1-2 is predicted also to be inactivated by loss of function of the tumour suppressor PTEN (phosphatase and tensin homologue deleted on chromosome 10) via activation of PKB, MAP4K3 may represent a promising new candidate for inhibition of the pathway in diseases of TSC1-2 or PTEN loss-of-function. Although the mechanism of MAP4K3 action on mTOR signalling has not been defined, the data on the positive regulation of both S6K1 activity and phosphorylation of 4E-BP1 by MAP4K3 overexpression points to the mTORC1 complex as the likely target of MAP4K3 action. Interestingly, addition of excess amino acids (relative to the normal concentrations found in DMEM) has been reported to fully activate S6K in the absence of growth factors, which is similar to current findings with overexpressed MAP4K3, suggesting that MAP4K3 may be activated further by amino acid excess. Future studies will be required to clarify these points. However, having defined the action of a new protein kinase in the mTOR pathway, delineating the signals downstream of amino acids that activate MAP4K3, and determining its mechanism of action will likely shed further light on nutrient regulation of cell growth (Findley, 2007).
In conclusion, these data indicate that amino acids stimulate the activity of a Ste20-family kinase, MAP4K3, with maximal kinase activation occurring concordant with phosphorylation of the Thr389 site of S6K1. In HeLa cells MAP4K3 activity is required for amino acid-induced activation of S6K1 and mediates rapamycin-senstive signalling to two effectors of mTORC1, but is not itself stimulated by insulin or inhibited by rapamycin. Lastly, MAP4K3 promotes cell growth in human HeLa cells in culture in a similar manner to Rheb and mTORC1 (Findley, 2007).
FoxO transcription factors and TORC1 are conserved downstream effectors of Akt. This study unraveled regulatory circuits underlying the interplay between Akt, FoxO, and mTOR. Activated FoxO1 inhibits mTORC1 by TSC2-dependent and TSC2-independent mechanisms. First, FoxO1 induces Sestrin3 (Sesn3) gene expression. Sesn3, in turn, inhibits mTORC1 activity in Tsc2-proficient cells. Second, FoxO1 elevates the expression of Rictor, leading to increased mTORC2 activity that consequently activates Akt. In Tsc2-deficient cells, the elevation of Rictor by FoxO increases mTORC2 assembly and activity at the expense of mTORC1, thereby activating Akt while inhibiting mTORC1. FoxO may act as a rheostat that maintains homeostatic balance between Akt and mTOR complexes' activities. In response to physiological stresses, FoxO maintains high Akt activity and low mTORC1 activity. Thus, under stress conditions, FoxO inhibits the anabolic activity of mTORC1, a major consumer of cellular energy, while activating Akt, which increases cellular energy metabolism, thereby maintaining cellular energy homeostasis (Chen, 2010; see graphical abstract).
The mTOR pathway is the central regulator of cell size. External signals from growth factors and nutrients converge on the mTORC1 multi-protein complex to modulate downstream targets, but how the different inputs are integrated and translated into specific cellular responses is incompletely understood. Deregulation of the mTOR pathway occurs in polycystic kidney disease (PKD), where cilia (filiform sensory organelles) fail to sense urine flow because of inherited mutations in ciliary proteins. It was therefore investigated if cilia have a role in mTOR regulation. This study shows that ablation of cilia in transgenic mice results in enlarged cells when compared with control animals. In vitro analysis demonstrated that bending of the cilia by flow is required for mTOR downregulation and cell-size control. Surprisingly, regulation of cell size by cilia is independent of flow-induced calcium transients, or Akt. However, the tumour-suppressor protein Lkb1 localises in the cilium, and flow results in increased AMPK phosphorylation at the basal body. Conversely, knockdown of Lkb1 prevents normal cell-size regulation under flow conditions. These results demonstrate that the cilium regulates mTOR signalling and cell size, and identify the cilium-basal body compartment as a spatially restricted activation site for Lkb1 signalling (2010).
Mechanistic target of rapamycin complex 1 (mTORC1) integrates diverse environmental signals to control cellular growth and organismal homeostasis. In response to nutrients, Rag GTPases (see Drosophila RagA-B) recruit mTORC1 to the lysosome to be activated, but how Rags are regulated remains incompletely understood. This study shows that Sestrins (see Drosophila Sestrin) bind to the heterodimeric RagA/B-RagC/D GTPases, and function as guanine nucleotide dissociation inhibitors (GDIs) for RagA/B. Sestrin overexpression inhibits amino-acid-induced Rag guanine nucleotide exchange and mTORC1 translocation to the lysosome. Mutation of the conserved GDI motif creates a dominant-negative form of Sestrin that renders mTORC1 activation insensitive to amino acid deprivation, whereas a cell-permeable peptide containing the GDI motif inhibits mTORC1 signaling. Mice deficient in all Sestrins exhibit reduced postnatal survival associated with defective mTORC1 inactivation in multiple organs during neonatal fasting. These findings reveal a nonredundant mechanism by which the Sestrin family of GDIs regulates the nutrient-sensing Rag GTPases to control mTORC1 signaling (Peng, 2014).
The tumor suppressor p53 is activated upon genotoxic and oxidative stress and in turn inhibits cell proliferation and growth through induction of specific target genes. Cell growth is positively regulated by mTOR, whose activity is inhibited by the TSC1:TSC2 complex. Although genotoxic stress has been suggested to inhibit mTOR via p53-mediated activation of mTOR inhibitors, the precise mechanism of this link was unknown. This study demonstrates that the products of two p53 target genes, Sestrin1 and Sestrin2 (see Drosophila Sestrin), activate the AMP-responsive protein kinase (AMPK) and target it to phosphorylate TSC2 and stimulate its GAP activity, thereby inhibiting mTOR. Correspondingly, Sestrin2-deficient mice fail to inhibit mTOR signaling upon genotoxic challenge. Sestrin1 and Sestrin2 therefore provide an important link between genotoxic stress, p53 and the mTOR signaling pathway (Budanov, 2008).
The mTOR signaling pathway is a central regulator of cell growth and survival. It is therefore not surprising that adverse environmental conditions negatively regulate cell growth by inhibiting mTOR. In addition to nutrient limitation, mTOR activity is negatively regulated by genotoxic stress and hypoxia, conditions that activate tumor suppressor p53. The ability of p53 to inhibit mTOR signaling is in line with its function as a negative regulator of cell growth and proliferation. The results described above strongly suggest that the ability of p53 to inhibit mTOR signaling depends on two of its target genes: Sesn1 and Sesn2 (Budanov, 2008).
The Sestrins belong to a small and evolutionary conserved family composed of 3 members in mammals, of which Sesn1 and 2 are stress inducible and p53 regulated. The ability of Sesn1/2 to inhibit cell growth and proliferation was attributed to their redox activity. The present work, however, demonstrates that Sesn1/2 are potent inhibitors of mTOR signaling, acting in a manner that does not depend on their redox activity, which only makes a partial contribution to their growth inhibitory activity. Sesn1 and 2 inhibit TORC1 activity towards p70S6K and 4E-BP1 in a variety of human and mouse cell lines, as well as in mouse liver. Notably, the ability of the hepatocarcinogen DEN to inhibit S6 phosphorylation is restricted to zone 3 hepatocytes, which are the main site in which it undergoes metabolic activation to become a potent alkylating agent, and this inhibitory activity is Sesn2-dependent. By inhibiting 4E-BP1 phosphorylation, Sesn2 enhances its interaction with eIF-4E and inhibits expression of growth regulatory proteins, such as cyclin D1 and c-Myc, whose translation is eIF-4E-dependent and sensitive to 4E-BP1 phosphorylation (Budanov, 2008).
The Sestrins impact TORC1 activity through the TSC1:TSC2 complex. Being a GAP for Rheb, the direct activator of TORC1, the TSC1:TSC2 complex is a central regulator of mTOR signaling. Sesn2 expression decreases Rheb GTP loading and the ability of both Sesn1 and Sesn2 to inhibit mTOR signaling is TSC2-dependent. One way to regulate TSC1:TSC2 GAP activity is through TSC2 phosphorylation, but other modes of regulation may also exist. Although the Sestrins have no effect on ERK and its target RSK or GSK3β, which can all serve as TSC2 kinases, they stimulate the activity of AMPK, a major TSC2 kinase. Furthermore, Sestrin expression enhanced TSC2 phosphorylation in live cells and this effect required the N-terminus of Sesn2, which mediates AMPKα binding. Sesn2 did not stimulate TSC1 phosphorylation and Sesn2-activated AMPK did not phosphorylate TSC1 (Budanov, 2008).
Importantly, the mTOR inhibitory activity of Sesn1/2 depends on AMPKα, whose phosphorylation at the activation loop was enhanced upon Sestrin expression. Inhibition of AMPK using compound C as well as shRNA silencing of AMPKα1 attenuated the ability of Sesn2 to inhibit mTOR signaling. Co-immunoprecipitation and gel filtration analyses revealed an interaction between Sesn2 and AMPKα, suggesting that Sestrins are engaged in formation of a large protein complex containing AMPK and TSC1:TSC2. It is proposed that Sesn1/2 induction in response to genotoxic stress results in binding of Sestrins, most likely as dimers, to AMPK and TSC1:TSC2, as well as auto-activation of AMPK through a mechanism based on induced proximity. In addition to activation of AMPK the Sestrins recruit it to phosphorylate TSC2. Phosphorylation of TSC2 correlates with enhancement of its GAP activity that leads to inhibition of Rheb and mTOR (Budanov, 2008).
Importantly, ample and clear evidence was obtained that Sesn1/2 are critical mediators of p53's ability to inhibit mTOR signaling. Using shRNA-mediated silencing it was found that both Sesn1 and Sesn2 participate in mTOR inhibition upon p53 activation in human cancer cells. Furthermore, disruption of the Sesn2 gene in mice attenuated the inhibition of p70S6K activity by the DNA-damaging agents: camptothecin in fibroblasts and DEN in hepatocytes. In both cases inhibition of p70S6K was p53-mediated, but unlike the p53 deficiency, the absence of Sesn2 has no effect on induction of p21Waf1, another p53 target gene. Thus, Sesn2 (and presumably Sesn1) seems to mediate only one aspect of p53 signaling -- inhibition of mTOR. Correspondingly, the growth-inhibitory activity of Sesn2 is not as strong as that of p53, which has additional targets with anti-proliferative activity, such as p21Waf1 (Budanov, 2008).
p53 deficiency and activation of mTOR signaling are hallmarks of human cancer. Several mechanisms account for mTOR activation in cancer, including activation of Ras, PI3K and AKT and inactivation of tumor suppressors that negatively regulate these molecules: PTEN, TSC1, TSC2 and LKB1. Although p53 can induce expression of several negative regulators of mTOR, including PTEN, TSC2, AMPKβ1 and IGF-BP3 in a cell type-dependent manner, the results demonstrate that p53-mediated inhibition of mTOR depends mainly on Sesn1 and 2 in mouse fibroblasts and certain human cancer cell lines and on Sesn2 in mouse liver (Budanov, 2008).
Inhibition of mTOR suppresses cell growth and proliferation. Sesn2 was known to inhibit cell proliferation, but its mechanism of action was heretofore unknown. The results strongly suggest that Sesn1 and Sesn2 exert their growth inhibitory effect via mTOR and may cooperate with other anti-proliferative p53 targets, such as p21Waf1. Interestingly, the SESN1 (6q21) and SESN2 (1p35) loci are frequently deleted in a variety of human cancers, suggesting they harbor one or more tumor-suppressors. Sesn2 deficiency was found to render murine fibroblasts more susceptible to oncogenic transformation and this effect may depend on mTOR inhibition. Hence, SESN1 and SESN2 may indeed be important components of the tumor suppressor network activated by p53 (Budanov, 2008).
In summary, while more remains to be learned about Sestrin biology and mechanism of action, the results establish these proteins as critical links between p53 and mTOR that enable p53 to inhibit cell growth (Budanov, 2008).
CBL and CBL-B ubiquitin ligases (see Drosophila Cbl) are negative regulators of tyrosine kinase signaling with established roles in the immune system. However, their physiological roles in epithelial tissues are unknown. This study used the MMTV-Cre-mediated Cbl gene deletion on a Cbl-b-null background as well as a tamoxifen-inducible mammary stem cell (MaSC)-specific Cbl/Cbl-b double knockout (DKO), using Lgr5-GFP-CreERT, to demonstrate a mammary epithelial cell-autonomous requirement of CBL and CBL-B in the maintenance of MaSCs. Using a newly engineered tamoxifen (TAM)-inducible Cbl/Cbl-b deletion model with a dual fluorescent reporter (Cblflox/flox; Cbl-bflox/flox; Rosa26-CreERT; mT/mG), it was shown that Cbl/Cbl-b DKO in mammary organoids leads to hyper-activation of AKT-mTOR signaling with depletion of MaSCs. Chemical inhibition of AKT or mTOR rescued MaSCs from Cbl/Cbl-b DKO induced depletion. These studies reveal a novel, cell-autonomous, requirement of CBL and CBL-B in epithelial stem cell maintenance during organ development and remodeling through modulation of mTOR signaling (Mohapatra, 2017).
The failure of axons to regenerate is a major obstacle for functional recovery after central nervous system (CNS) injury. Removing extracellular inhibitory molecules results in limited axon regeneration in vivo. To test for the role of intrinsic impediments to axon regrowth, cell growth control genes were analyzed using a virus-assisted in vivo conditional knockout approach. Deletion of PTEN (phosphatase and tensin homolog), a negative regulator of the mammalian target of rapamycin (mTOR) pathway, in adult retinal ganglion cells (RGCs) promotes robust axon regeneration after optic nerve injury. In wild-type adult mice, the mTOR activity is suppressed and new protein synthesis is impaired in axotomized RGCs, which may contribute to the regeneration failure. Reactivating this pathway by conditional knockout of tuberous sclerosis complex 1, another negative regulator of the mTOR pathway, also leads to axon regeneration. Thus, these results suggest the manipulation of intrinsic growth control pathways as a therapeutic approach to promote axon regeneration after CNS injury (Park, 2008).
The mTORC1 and mTORC2 pathways regulate cell growth, proliferation, and survival. This study identified DEPTOR as an mTOR-interacting protein whose expression is negatively regulated by mTORC1 and mTORC2. The gene for DEPDC6 is found only in vertebrates, and encodes a protein with tandem N-terminal DEP (dishevelled, egl-10, pleckstrin) domains and a C-terminal PDZ (postsynaptic density 95, discs large, zonula occludens-1) domain. Loss of DEPTOR activates S6K1, Akt, and SGK1, promotes cell growth and survival, and activates mTORC1 and mTORC2 kinase activities. DEPTOR overexpression suppresses S6K1 but, by relieving feedback inhibition from mTORC1 to PI3K signaling, activates Akt. Consistent with many human cancers having activated mTORC1 and mTORC2 pathways, DEPTOR expression is low in most cancers. Surprisingly, DEPTOR is highly overexpressed in a subset of multiple myelomas harboring cyclin D1/D3 or c-MAF/MAFB translocations. In these cells, high DEPTOR expression is necessary to maintain PI3K and Akt activation and a reduction in DEPTOR levels leads to apoptosis. Thus, this study identified a novel mTOR-interacting protein whose deregulated overexpression in multiple myeloma cells represents a mechanism for activating PI3K/Akt signaling and promoting cell survival (Peterson, 2009).
Loss-of-function data indicate that DEPTOR inhibits both the mTORC1 and mTORC2 pathways. However, by inhibiting mTORC1, DEPTOR overexpression relieves mTORC1-mediated inhibition of PI3K, causing an activation of PI3K and, paradoxically, of mTORC2-dependent outputs, like Akt (Peterson, 2009).
mTOR interacts with DEPTOR via its PDZ domain, and so far there is no information about the function of the tandem DEP domains the protein also contains. In other proteins, DEP domains mediate protein-protein interactions, but in numerous DEPTOR purifications additional DEPTOR-interacting proteins were not identified, besides the known components of mTORC1 and mTORC2. Therefore, based on current evidence, DEPTOR appears dedicated to mTOR regulation, and it is proposed that in vertebrates it is likely to be involved in regulating other outputs of the mTOR signaling network besides the growth and survival pathways examined in this study. The mTOR complexes and DEPTOR negatively regulate each other, suggesting the existence of a feedforward loop in which the loss of DEPTOR leads to an increase in mTOR activity, which then further reduces DEPTOR expression. This type of regulatory circuit should result in DEPTOR expression being tightly coupled to mTOR activity, and, interestingly, it was noted that DEPTOR mRNA levels strongly anticorrelate with cell size, a readout of mTORC1 activity (Peterson, 2009).
About 28% of human multiple myelomas (MMs) overexpress DEPTOR. These results are consistent with a published survey of 67 MM tumors and 43 MM cell lines, in which 21% were shown to possess copy number gains and associated expression increases of the genes within a 6 Mb region of chromosome 8q24 that contains DEPTOR. Furthermore, it appears that deregulated overexpression of c-MAF and MAFB is an additional, perhaps even more prevalent, mechanism for increasing DEPTOR expression in MMs. The related c-MAF and MAFB transcription factors are expressed (frequently as the result of chromosomal translocations) in a large fraction of MMs, but not in the plasma cells from which they are derived. Consistent with c-MAF playing a key role in promoting DEPTOR expression, a knockdown of c-MAF in a MM cell line having a c-MAF translocation decreases the expression of DEPTOR and mimics the effects of a DEPTOR knockdown on mTOR and PI3K signaling. The levels of the DEPTOR and c-MAF or MAFB mRNAs highly correlate with each other and, importantly, DEPTOR expression correlates with poor survival in patients with multiple myeloma (Peterson, 2009).
In many multiple myeloma cell lines, DEPTOR is massively overexpressed compared to the levels found in other cancer cell lines, such as HeLa cells. In these cells, the great overexpression of DEPTOR inhibits mTORC1 growth signaling and drives outputs dependent on PI3K. Interestingly, a reduction in DEPTOR expression to the lower levels seen in non-multiple myeloma cell lines causes cell death via apoptosis. This suggests that a pharmacologically induced reduction in DEPTOR expression or disruption of the DEPTOR-mTOR interaction could have therapeutic benefits for the treatment of multiple myeloma. There has been progress in developing small-molecule inhibitors of protein-protein interactions mediated by PDZ domains, so it is conceivable that blockers of the DEPTOR-mTOR interaction could be made (Peterson, 2009).
Although a number of other cancer cell lines have high levels of DEPTOR, as a class only multiple myelomas appear to consistently overexpress it. Besides activating PI3K/Akt signaling, DEPTOR overexpression in MM cells may provide these cells with benefits that are not relevant in other cancer types or perhaps even detrimental. For example, the high demand that MM cells place on the protein synthesis machinery to produce large amounts of immunoglobulins, causes a significant ER stress, which renders these cells susceptible to apoptosis induction via agents that induce further ER stress, such as proteasome inhibitors. DEPTOR overexpression, by partial inhibition of protein synthesis through the suppression of mTORC1, may reduce the levels of ER stress below the threshold that triggers apoptosis. In contrast, in other cancer cells in which ER stress is not a significant factor, DEPTOR overexpression may be selected against because reduced rates of protein synthesis may not be tolerated. That mTORC1-stimulated protein synthesis leads to ER stress is already appreciated as TSC1 or TSC2 null cells have increased sensitivity to ER stress-induced death (Peterson, 2009).
It is curious that DEPTOR is overexpressed mostly in MMs characterized by chromosomal translocations instead of those that are hyperdiploid because of aneuploidy. Elevated DEPTOR expression might be tolerated better in the nonhyperdiploid MMs because aneuploidy itself increases sensitivity to conditions, like mTORC1 inhibition, that interfere with protein synthesis. Moreover, the state of high mTORC2 and low mTORC1 signaling that this work indicates that some MM cells prefer cannot be achieved by mutations that activate PI3K signaling, perhaps explaining why multiple myelomas exhibit low rates of PTEN-inactivating or PI3K-activating mutations (Peterson, 2009).
Amino acids are required for activation of the mammalian target of rapamycin (mTOR) kinase which regulates protein translation, cell growth, and autophagy. Cell surface transporters that allow amino acids to enter the cell and signal to mTOR are unknown. This study shows that cellular uptake of L-glutamine and its subsequent rapid efflux in the presence of essential amino acids (EAA) is the rate-limiting step that activates mTOR. L-glutamine uptake is regulated by SLC1A5 and loss of SLC1A5 function inhibits cell growth and activates autophagy. The molecular basis for L-glutamine sensitivity is due to SLC7A5/SLC3A2, a bidirectional transporter that regulates the simultaneous efflux of L-glutamine out of cells and transport of L-leucine/EAA into cells. Certain tumor cell lines with high basal cellular levels of L-glutamine bypass the need for L-glutamine uptake and are primed for mTOR activation. Thus, L-glutamine flux regulates mTOR, translation and autophagy to coordinate cell growth and proliferation (Nicklin, 2009).
Autophagy is a conserved cellular process to degrade and recycle cytoplasmic components. During autophagy, lysosomes fuse with an autophagosome to form an autolysosome. Sequestered components are degraded by lysosomal hydrolases and presumably released into the cytosol by lysosomal efflux permeases. Following starvation-induced autophagy, lysosome homeostasis is restored by autophagic lysosome reformation (ALR) requiring activation of the 'target of rapamycin' (TOR) kinase. Spinster (Spin) encodes a putative lysosomal efflux permease with the hallmarks of a sugar transporter. Drosophila spin mutants accumulate lysosomal carbohydrates and enlarged lysosomes. This study shows that defects in spin, in both mammalian cells and Drosophila, lead to the accumulation of enlarged autolysosomes. spin is essential for mTOR reactivation and lysosome reformation following prolonged starvation. Further, the sugar transporter activity of Spin is essential for ALR (Rong, 2011).
During autophagy, lysosomes fuse with autophagosomes to form autolysosomes, where contents are degraded by lysosomal hydrolases and released by lysosomal efflux transporters. The autophagic/lysosomal pathway is critical to cellular homeostasis. Defects in autophagy lead to the accumulation of damaged organelles, misfolded proteins, and toxic metabolites, and are associated with neurodegeneration and other abnormalities. Defects in specific lysosomal hydrolysis have been implicated in lysosomal storage disorders (LSDs). Loss of lysosomal protease activity can lead to the accumulation of undigested material, as well as neurodegenerative disease. In addition, defective efflux of lysosomal contents by lysosomal transporters can lead to accumulation of lysosomal substrates and defective lysosomal function (Rong, 2011).
Lysosomal efflux transporters are a family of lysosomal membrane proteins required for the export of lysosomal degradation products, such as amino acids and monosaccharides. A subset of lysosomal storage diseases has been linked to mutations found in lysosomal efflux transporters. For example, defects in Sialin, a sialic acid transporter, leads to sialic acid storage diseases (SASD), and defects in the lysosome Arginine transporter lead to Juvenile Batten Disease. Spinster (Spin) (also known as benchwarmer) is a late endosomal/lysosomal membrane protein with the amino acid sequence of a lysosomal sugar carrier in the major facilitator superfamily. Spin is a transmembrane protein containing 8-12 transmembrane domains. In Drosophila, hypomorphic mutations in spin lead to decreased adult life span, defects in courtship behavior, accumulation of autoflourescent pigments, and neurodegeneration. Drosophila spin mutants also exhibit neuromuscular synaptic overgrowth and enhanced tau-mediated toxicity. In zebrafish, loss of the spin homolog not really started (nrs) leads to embryonic lethality characterized by the accumulation of opaque substances in the yolk. Interestingly, Drosophila spin mutants exhibit endocytic defects, as well as widespread accumulation of lysosomal carbohydrates and enlarged lysosomes. Little is known, however, about the mechanism leading to the accumulation of enlarged lysosomes in spin mutants (Rong, 2011).
ALR is an evolutionarily conserved lysosome regeneration cycle that governs nutrient sensing and lysosome homeostasis following starvation-induced autophagy. In response to starvation, mTOR is inhibited, leading to the induction of autophagy. After prolonged starvation, however, mTOR is reactivated. Upon mTOR reactivation, tubules extrude from autolysosomal membranes and give rise to vesicles that ultimately mature into functional lysosomes. The degradation of autophagic cargo is required for mTOR reactivation after starvation, and inhibiting mTOR reactivation leads to the accumulation of enlarged autolysosomes. In addition, ALR requires the dissociation of the small GTPase Rab7 from autolysosomes, and overexpression of constitutively active Rab7 results in the accumulation of enlarged autolysosomes (Rong, 2011).
This study reports that loss of spin leads to the accumulation of enlarged autolysosomes that fail to degrade their contents in both mammalian cells and Drosophila. spin is required for mTOR reactivation and lysosome reformation following prolonged starvation. Interestingly, reactivation of mTOR signaling after starvation is sufficient to induce lysosome reformation even in the context of decreased spin function. Importantly, it was found that the sugar transporter activity of spin is essential for ALR. These findings elucidate the role of this lysosomal efflux transporter in ALR and reveal its contribution to LSDs (Rong, 2011).
Drosophila spin mutants have been shown to exhibit progressive neurodegeneration, and lysosomal abnormalities have been shown to contribute to neurodegeneration in this context. Nonetheless, how defects in spin lead to lysosomal abnormalities has been unclear. This study demonstrates that the structures that accumulate in spin mutants are autolysosomes, and that spin is required for ALR following prolonged starvation (Rong, 2011).
Abnormalities in lysosome function and morphology become apparent only under starvation conditions in cultured mammalian cells. This might be explained by the fact that under nutrient-rich conditions, the influx of lysosome cargo is limited, but when cells undergo autophagy, lysosome cargo influx increases, magnifying the severity of the defect. If this hypothesis is correct, then one must wonder why spin mutant flies exhibit an accumulation of slightly enlarged lysosomal-associated membrane protein 1-(Lamp1-) positive structures even when fed. One possibility is that the metabolism of flies in this respect is greater than the metabolism of mammals. Another possible explanation is that the fluctuation of nutrients in vivo is much greater than in cells grown in culture medium. Thus, a nonstarved animal could have higher basal levels of autophagy compared with cells maintained in culture. This might be particularly important in the brain because selective deletion of Atg5 in neuronal cells leads to neurodegeneration even in unstarved mice (Rong, 2011).
One question is how Spin, a lysosomal efflux sugar transporter, alters the protein degradation capacity of lysosomes. Lysosome protein degradation capacity is dependent on the lysosomal internal environment. Lysosome pH is one of the major factors regulating lysosomal degradation ability, as the optimal pH for many lysosomal proteases is around 4.5; in either lower or higher pH, lysosome protease activity is compromised. Interestingly, it was found that upon starvation, spin knock-down cells exhibit a dramatically decreased lysosomal pH. However, it is unknown how spin knock-down leads to an increase in lysosomal acidity. One possibility is that spin is a H+/sugar symporter. H+/amino acid symporters have been identified in lysosomes, for example, LYAAT1, a lysosome amino acid efflux transporter, is a H+/amino acid symporter, and the efflux transport of amino acids by LYAAT1 is driven by the H+ gradient. Similarly, cystinosin, a lysosomal cystine efflux transporter which also contains seven transmembrane domains, has been reported to be a H+/cystine symporter which uses H+ to drive cystine efflux transport. A recent structural study for FucP, a H+/Fucose symporter which also belongs to the major facilitator superfamily (MFS) of transporters further supports this hypothesis. A structural study showed that a conserved E residue must be protonated for proper Fucose transport. Interestingly, in a rescue experiment carried out in this stuyd, the E217K mutation failed to rescue ALR in spin knockdown cells. If this hypothesis is correct, it would be expected that spin knockdown would block both sugar and H+ efflux and lead to lysosomal acidification, and autolysosomal degradation defects. Further investigation will be required to explore this possibility (Rong, 2011).
There is a bidirectional regulation between autolysosomal degradation and mTOR reactivation/ALR. On one hand, degradation of autolysosomal content is required for mTOR reactivation and ALR; on the other hand, defective mTOR reactivation/ALR also causes the impairment of degradation. For example, if mTOR reactivation/ALR is blocked during starvation by either adding the mTOR inhibitor rapamycin or knocking down mTOR, the degradation of autophagy substrate LC3 is impaired. Interestingly, this study found that adding fetal calf serum rapidly, albeit partially, restores the pH and degradation capacity of spin knockdown cells, indicating that mTOR may play a role in regulating the pH, and thus the degradation capacity, of autolysosomes. These data raise the interesting possibility that mTOR may regulate ALR by affecting the degradation capacity of autolysosomes (Rong, 2011).
One important implication of the current findings is that ALR may play an important role in disease progression in LSDs. The phenotype of spin mutant flies has been long considered to be similar to certain LSDs. This study demonstrated that a spin knockdown also leads to defects in autolysosomal degradation and ALR. These data clearly demonstrate that ALR is important to the maintenance of lysosome-based cellular degradation capacity. The basal level of autophagy may be higher in vivo than cultured cells due to the greater fluctuation of nutrients during the feeding cycle. Thus, it is conceivable that a defect in ALR could cause LSD phenotypes in fed animals (Rong, 2011).
A long-lasting question in LSDs is why a mutation in a single lysosomal enzyme can cause overall lysosome degradation failure. Because the regulation between lysosomal degradation and ALR is bidirectional, the data suggest the interesting possibility that a minor defect in lysosomal degradation capacity could cause a minor defect in ALR, which could in turn amplify the lysosomal/autolysosomal degradation defect. The positive feedback nature of this regulation loop then may eventually cause the progressive pathological features of LSDs (Rong, 2011).
p70 S6 kinase alpha (p70alpha) is activated in vivo through a multisite phosphorylation in response to mitogens if a sufficient supply of amino acids is available or in response to high concentrations of amino acids per se. The immunosuppressant drug rapamycin inhibits p70alpha activation in a manner that can be overcome by coexpression of p70alpha with a rapamycin-resistant mutant of the mammalian target of rapamycin (mTOR) but only if the mTOR kinase domain is intact. A mammalian recombinant p70alpha polypeptide, extracted in an inactive form from rapamycin-treated cells, can be directly phosphorylated by the mTOR kinase in vitro predominantly at the rapamycin-sensitive site Thr-412. mTOR-catalyzed p70alpha phosphorylation in vitro is accompanied by a substantial restoration in p70alpha kinase activity toward its physiologic substrate, the 40 S ribosomal protein S6. Moreover, sequential phosphorylation of p70alpha by mTOR and 3-phosphoinositide-dependent protein kinase 1 in vitro results in a synergistic stimulation of p70alpha activity to levels similar to those attained by serum stimulation in vivo. These results indicate that mTOR is likely to function as a direct activator of p70 in vivo, although the relative contribution of mTOR-catalyzed p70 phosphorylation in each of the many circumstances that engender p70 activation remains to be defined (Isotani, 2000).
PDK1-catalyzed phosphorylation of Thr-252 on the p70alpha activation loop is a critical and probably final step in the physiologic activation of p70alpha in vivo. The ability of PDK1 to phosphorylate Thr-252 is regulated primarily by the accessibility of the p70alpha activation loop to PDK1, which in turn is controlled by a series of prior p70alpha phosphorylations. Phosphorylation of the multiple Ser/Thr-Pro motifs clustered in the pseudosubstrate autoinhibitory segment of the p70alpha carboxyl-terminal tail serves to disocclude the catalytic domain, greatly enhancing access to PDK1. A similar effect can be achieved by deletion of the p70alpha carboxyl-terminal tail (to give p70alpha-DeltaCT104). At any level of PDK1 activity, the extent of Thr-252 phosphorylation of p70alpha-DeltaCT104 is substantially greater than with a similar amount of full-length p70alpha polypeptide. In addition, the S6 kinase activity generated by any extent of PDK1-catalyzed Thr-252 phosphorylation is significantly higher for p70alpha-DeltaCT104 as compared with full-length p70alpha. Displacement of the p70alpha carboxyl-terminal tail is also necessary for the phosphorylation of Thr-412 in vivo, and modification of Thr-412 itself significantly enhances the ability of PDK1 to phosphorylate Thr-252. In addition, the simultaneous phosphorylation of Thr-412 and Thr-252 appears to generate a synergistic activation of p70alpha. Thus, the substitution of Thr-412 by Glu in p70alpha-DeltaCT104 alone gives a 6-fold increase in S6 kinase activity, and the PDK1 catalyzed phosphorylation of p70alpha-DeltaCT104 Thr-252 alone gives a 15-fold increase, but the two modifications together give at least a 240-fold increase in S6 kinase activity over the unmodified p70alpha-DeltaCT104 polypeptide. The importance of the strong positively cooperative effect of Thr-252 and Thr-412 phosphorylation for the physiologic activation of p70alpha is illustrated by the response of p70alpha to the inhibitors rapamycin and wortmannin; these agents each cause a rapid dephosphorylation of Thr-412 but a slower and lesser dephosphorylation of Thr-252. Despite the preservation of Thr-252 phosphorylation, S6 kinase activity in the presence of rapamycin or wortmannin declines in parallel to Thr-412 dephosphorylation. In view of the ability of mTOR to catalyze the in vitro phosphorylation of p70alpha Thr-412 as well as sites within the autoinhibitory segment in the p70alpha carboxyl-terminal tail and the potential effects of such phosphorylations on the response of p70alpha to PDK1, the p70alpha activation achieved in vitro by mTOR or PDK1 alone have been compared to that achieved by sequential phosphorylation by mTOR and PDK1 and to that achieved in vivo by stimulation of the cells with 10% serum. mTOR alone increases the S6 kinase activity of p70alpha in vitro by more than 10-fold, whereas PDK1 alone hardly activates p70alpha, presumably reflecting the relatively poor access of Thr-252 to PDK1 in full-length, inactive p70alpha. In contrast, phosphorylation of p70alpha by PDK1 after a prior phosphorylation by mTOR increases the p70alpha activity by 10-fold over that engendered by mTOR alone, to a level roughly 70-fold greater than that generated by PDK1 acting alone. Moreover, the S6 kinase activity generated in vitro by the sequential action of mTOR and PDK1 is indistinguishable from that achieved in vivo by stimulation of cells with 10% serum (Isotani, 2000).
The mammalian target of rapamycin (mTOR) controls the translation machinery via activation of S6 kinases 1 and 2 (S6K1/2) and inhibition of the eukaryotic initiation factor 4E (eIF4E) binding proteins 1, 2, and 3 (4E-BP1/2/3). S6K1 and 4E-BP1 are regulated by nutrient-sensing and mitogen-activated pathways. The molecular basis of mTOR regulation of S6K1 and 4E-BP1 remains controversial. A conserved TOR signaling (TOS) motif has been identified in the N terminus of all known S6 kinases and in the C terminus of the 4E-BPs; the TOS motif is crucial for phosphorylation and regulation S6K1 and 4E-BP1 activities. Deletion or mutations within the TOS motif significantly inhibit S6K1 activation and the phosphorylation of its hydrophobic motif, Thr389. In addition, this sequence is required to suppress an inhibitory activity mediated by the S6K1 C terminus. The TOS motif is essential for S6K1 activation by mTOR, since mutations in this motif mimic the effect of rapamycin on S6K1 phosphorylation, and render S6K1 insensitive to changes in amino acids. Furthermore, overexpression of S6K1 with an intact TOS motif prevents 4E-BP1 phosphorylation by a common mTOR-regulated modulator of S6K1 and 4E-BP1. It is concluded that S6K1 and 4E-BP1 contain a conserved five amino acid sequence (TOS motif) that is crucial for their regulation by the mTOR pathway. mTOR seems to regulate S6K1 by two distinct mechanisms. The TOS motif appears to function as a docking site for either mTOR itself or a common upstream activator of S6K1 and 4E-BP1 (Schalm, 2002).
Neuropoletic cytokines such as ciliary neurotrophic factor (CNTF) can activate multiple signaling pathways in parallel, including those involving Janus kinase (JAK)-signal transducers and activators of transcription (STATs), mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI 3-kinase) and mammalian target of rapamycin (mTOR)-p70 S6 kinase. Crosstalk occurs between these pathways, because studies have shown that STAT3 requires phosphorylation on tyrosine and serine residues by independent protein kinase activities for maximal activation of target gene transcription. Members of the JAK/Tyk family of tyrosine kinases mediate phosphorylation of STAT3 at Tyr705 during CNTF signaling; however, the kinase responsible for phosphorylation at STAT3 Tyr727 appears to depend on both the extracellular stimulus and the cellular context. This study investigates the kinase activity responsible for phosphorylation of STAT3 on Ser727 in CNTF-stimulated neuroblastoma cells. CNTF-induced phosphorylation of Ser727 is inhibited by the mTOR inhibitor rapamycin, but not by inhibitors of MAPK and protein kinase C (PKC) activation. A STAT3 peptide is efficiently phosphorylated on Ser727 in a CNTF-dependent manner by mTOR, but not by a kinase-inactive mTOR mutant or by p70 S6 kinase. In agreement with these biochemical studies, rapamycin treatment of cells transfected with a STAT-responsive promoter reporter decreases activation of the reporter to the same degree as a STAT3 Ser727Ala mutant. The ability of mTOR to contribute to activation of STAT3 extends the function of mTOR in mammalian cells to include transcriptional regulation (Yokogami, 2000).
Under serum-free conditions, rapamycin, an inhibitor of mammalian target of rapamycin (mTOR), induces apoptosis of cells lacking functional p53. Cells expressing wild-type p53 or p21Cip1 arrest in G1 and remain viable. In cells lacking functional p53, rapamycin or amino acid deprivation induces rapid and sustained activation of apoptosis signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinase, and elevation of phosphorylated c-Jun that results in apoptosis. This stress response depends on expression of eukaryotic initiation factor 4E binding protein 1 and is suppressed by p21Cip1 independent of cell cycle arrest. Rapamycin induces p21Cip1 binding to ASK1, suppressing kinase activity and attenuating cellular stress. These results suggest that inhibition of mTOR triggers a potentially lethal response that is prevented only in cells expressing p21Cip1 (Huang, 2003).
In cycling cells, transcription of ribosomal RNA genes by RNA polymerase I (Pol I) is tightly coordinated with cell growth. The mammalian target of rapamycin (mTOR) regulates Pol I transcription by modulating the activity of TIF-IA, a regulatory factor that senses nutrient and growth-factor availability. Inhibition of mTOR signaling by rapamycin inactivates TIF-IA and impairs transcription-initiation complex formation. Moreover, rapamycin treatment leads to translocation of TIF-IA into the cytoplasm. Rapamycin-mediated inactivation of TIF-IA is caused by hypophosphorylation of Ser 44 (S44) and hyperphosphorylation of Ser 199 (S199). Phosphorylation at these sites affects TIF-IA activity in opposite ways, for example, phosphorylation of S44 activates and S199 inactivates TIF-IA. The results identify a new target for mTOR-signaling pathways and elucidate the molecular mechanism underlying mTOR-dependent regulation of rRNA synthesis (Mayer, 2004).
An important question is whether mTOR itself, or a downstream kinase, phosphorylates TIF-IA. S6K1 is known to act downstream of mTOR, raising the possibility that S6K1 directly phosphorylates TIF-IA. However, TIF-IA contains no consensus recognition motif for S6K1, and neither immunopurified mTOR nor S6K1 were capable of phosphorylating TIF-IA in vitro. Ser 44 resides within a recognition motif for Cdks (SPxR). This raises the possibility that Cdk-mediated phosphorylation of S44 links nutrient-sensing pathways to cellcycle control. Pol I transcription oscillates during the cell cycle, being highest in S2 and G2 phase, shutting down at mitosis and recovering during G1-phase progression. It is not yet known whether or not TIF-IA is inactivated during mitosis and is reactivated by phosphorylation by G1-specific Cdks. The finding that immunopurified Cdk2/cyclin E phosphorylates S44 and phosphorylation at this site is required for TIF-IA function, supports the view that phosphorylation by Cdks is essential for regulation of TIF-IA activity and, hence, cell cycle-dependent fluctuations of Pol I transcription. In contrast, although rapamycin inhibits Cdk activation and compromises cell-cycle progression, inhibition of S44 phosphorylation by G1-phase Cdks is unlikely to be responsible for the rapid switch-off of pre-rRNA synthesis, because TIF-IA inactivation was observed as early as 1 h after rapamycin administration. Inactivation of TIF-IA by rapamycin has not been observed in the presence of protein phosphatase inhibitors, indicating that activation of phosphatase(s) rather than inhibition of Cdks induces rapamycin-dependent hypophosphorylation of S44. In support of this, PP2A is very rapidly activated by rapamycin, and the regulated activity of PP2A has been proposed as a master switch to regulate the activity of downstream effectors of mTOR, such as S6K1 and 4E-BP. Like TIF-IA, S6K1 and 4E-BP are phosphorylated at multiple sites and are dephosphorylated within 30 min after rapamycin treatment (Mayer, 2004).
While NF-kappaB is considered to play key roles in the development and progression of many cancers, the mechanisms whereby this transcription factor is activated in cancer are poorly understood. A key oncoprotein in a variety of cancers is the serine- threonine kinase Akt, which can be activated by mutations in PI3K, by loss of expression/activity of PTEN, or through signaling induced by growth factors and their receptors. A key effector of Akt-induced signaling is the regulatory protein mTOR (mammalian target of rapamycin). This study shows that mTOR downstream from Akt controls NF-kappaB activity in PTEN-null/inactive prostate cancer cells via interaction with and stimulation of IKK. The mTOR-associated protein Raptor is required for the ability of Akt to induce NF-kappaB activity. Correspondingly, the mTOR inhibitor rapamycin is shown to suppress IKK activity in PTEN-deficient prostate cancer cells through a mechanism that may involve dissociation of Raptor from mTOR. The results provide insight into the effects of Akt/mTOR-dependent signaling on gene expression and into the therapeutic action of rapamycin (Dan, 2008).
Radial glial cells (RCGs) are self-renewing progenitor cells that give rise to neurons and glia during embryonic development. Throughout neurogenesis, these cells contact the cerebral ventricles and bear a primary cilium. Although the role of the primary cilium in embryonic patterning has been studied, its role in brain ventricular morphogenesis is poorly characterized. Using conditional mutants, this study showed that the primary cilia of radial glia determine the size of the surface of their ventricular apical domain through regulation of the mTORC1 pathway. In cilium-less mutants, the orientation of the mitotic spindle in radial glia is also significantly perturbed and associated with an increased number of basal progenitors. The enlarged apical domain of RGCs leads to dilatation of the brain ventricles during late embryonic stages (ventriculomegaly), which initiates hydrocephalus during postnatal stages. These phenotypes can all be significantly rescued by treatment with the mTORC1 inhibitor rapamycin. These results suggest that primary cilia regulate ventricle morphogenesis by acting as a brake on the mTORC1 pathway. This opens new avenues for the diagnosis and treatment of hydrocephalus (Foerster, 2017)
The mammalian target of rapamycin, mTOR, is a central regulator of cell growth. Its activity is regulated by Rheb, a Ras-like small guanosine triphosphatase (GTPase), in response to growth factor stimulation and nutrient availability. Rheb regulates mTOR through FKBP38, a member of the FK506-binding protein (FKBP) family that is structurally related to FKBP12. FKBP38 binds to mTOR and inhibits its activity in a manner similar to that of the FKBP12-rapamycin complex. Rheb interacts directly with FKBP38 and prevents its association with mTOR in a guanosine 5'-triphosphate (GTP)-dependent manner. These findings suggest that FKBP38 is an endogenous inhibitor of mTOR, whose inhibitory activity is antagonized by Rheb in response to growth factor stimulation and nutrient availability (Bai, 2007).
mTOR controls cell growth, in part by regulating p70 S6 kinase alpha (p70alpha) and eukaryotic initiation factor 4E binding protein 1 (4EBP1). Raptor is a 150 kDa mTOR binding protein that also binds 4EBP1 and p70alpha. The binding of raptor to mTOR is necessary for the mTOR-catalyzed phosphorylation of 4EBP1 in vitro, and it strongly enhances the mTOR kinase activity toward p70alpha. Rapamycin or amino acid withdrawal increases, whereas insulin strongly inhibits, the recovery of 4EBP1 and raptor on 7-methyl-GTP Sepharose. Partial inhibition of raptor expression by RNA interference (RNAi) reduces mTOR-catalyzed 4EBP1 phosphorylation in vitro. RNAi of C. elegans raptor yields an array of phenotypes that closely resemble those produced by inactivation of Ce-TOR. Thus, raptor is an essential scaffold for the mTOR-catalyzed phosphorylation of 4EBP1 and mediates TOR action in vivo (Hara, 2002).
mTOR/RAFT1/FRAP is the target of the immunosuppressive drug rapamycin and the central component of a nutrient- and hormone-sensitive signaling pathway that regulates cell growth. mTOR forms a stoichiometric complex with raptor, an evolutionarily conserved protein with at least two roles in the mTOR pathway. Raptor has a positive role in nutrient-stimulated signaling to the downstream effector S6K1, maintenance of cell size, and mTOR protein expression. The association of raptor with mTOR also negatively regulates the mTOR kinase activity. Conditions that repress the pathway, such as nutrient deprivation and mitochondrial uncoupling, stabilize the mTOR-raptor association and inhibit mTOR kinase activity. It is proposed that raptor is a missing component of the mTOR pathway that through its association with mTOR regulates cell size in response to nutrient levels (Kim, 2002).
mTOR and raptor are components of a signaling pathway that regulates mammalian cell growth in response to nutrients and growth factors. A member of this pathway, a protein named GbetaL, has been identified that binds to the kinase domain of mTOR and stabilizes the interaction of raptor with mTOR. Like mTOR and raptor, GbetaL participates in the control of cell size and in nutrient- and growth factor-mediated signaling to S6K1, a downstream effector of mTOR. The binding of GbetaL to mTOR strongly stimulates the kinase activity of mTOR toward S6K1 and 4E-BP1, an effect reversed by the stable interaction of raptor with mTOR. Interestingly, nutrients and rapamycin regulate the association between mTOR and raptor only in complexes that also contain GbetaL. Thus, it is proposed that the opposing effects on mTOR activity of the GbetaL- and raptor-mediated interactions regulate the mTOR pathway (Kim, 2003).
The translational repressor protein eIF4E-binding protein 1 (4E-BP1, also termed PHAS-I) is regulated by phosphorylation through the rapamycin-sensitive mTOR (mammalian target of rapamycin) pathway. Recent studies have identified two regulatory motifs in 4E-BP1, an mTOR-signaling motif (TOS) in the C-terminus of 4E-BP1 and a RAIP motif (after its sequence) in the N-terminus. Other work has shown that the protein raptor binds to mTOR and 4E-BP1. Raptor binds to full-length 4E-BP1 or a C-terminal fragment containing the TOS motif but not to an N-terminal fragment containing the RAIP motif. Mutation of several residues within the TOS motif abrogates binding to raptor, indicating that the TOS motif is required for this interaction. 4E-BP1 undergoes phosphorylation at multiple sites in intact cells. The effects of removal or mutation of the RAIP and TOS motifs differ. The RAIP motif is absolutely required for phosphorylation of sites in the N- and C-termini of 4E-BP1, while the TOS motif primarily affects phosphorylation of Ser64/65 and Thr69/70, and also the rapamycin-insensitive site Ser101. Phosphorylation of N-terminal sites that are dependent upon the RAIP motif are sensitive to rapamycin. The RAIP motif thus promotes the mTOR-dependent phosphorylation of multiple sites in 4E-BP1 independently of the 4E-BP1/raptor interaction (Beugnet, 2003).
Mammalian target of rapamycin (mTOR) is the central element of a signaling pathway involved in the control of mRNA translation and cell growth. The actions of mTOR are mediated in part through the phosphorylation of the eukaryotic initiation factor 4E-binding protein, PHAS-I. In vitro mTOR phosphorylates PHAS-I in sites that control PHAS-I binding to eukaryotic initiation factor 4E; however, whether mTOR directly phosphorylates PHAS-I in cells has been a point of debate. The Arg-Ala-Ile-Pro (RAIP motif) and Phe-Glu-Met-Asp-Ile (tor signaling motif) sequences found in the NH2- and COOH-terminal regions of PHAS-I, respectively, are required for the efficient phosphorylation of PHAS-I in cells. Mutations in either motif markedly decrease the phosphorylation of recombinant PHAS-I by mTOR in vitro. Wild-type PHAS-I, but none of the mutant proteins, is coimmunoprecipitated with hemagglutinin-tagged raptor, an mTOR-associated protein, after extracts of cells overexpressing raptor have been supplemented with recombinant PHAS-I proteins. Moreover, raptor overexpression enhances the phosphorylation of wild-type PHAS-I by mTOR but not the phosphorylation of the mutant proteins. The results not only provide direct evidence that both the RAIP and tor signaling motifs are important for the phosphorylation by mTOR, possibly by allowing PHAS-I binding to raptor, but also support the view that mTOR phosphorylates PHAS-I in cells (Choi, 2003).
The mammalian target of rapamycin (mTOR) controls multiple cellular functions in response to amino acids and growth factors, in part by regulating the phosphorylation of p70 S6 kinase (p70S6k) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1; see Drosophila Thor). Raptor (regulatory associated protein of mTOR) also binds p70S6k and 4E-BP1 and is essential for TOR signaling in vivo. Raptor binds to p70S6k and 4E-BP1 through their respective TOS (conserved TOR signaling) motifs shown to be required for amino acid- and mTOR-dependent regulation of these mTOR substrates in vivo. A point mutation of the TOS motif also eliminates all in vitro mTOR-catalyzed 4E-BP1 phosphorylation and abolishes the raptor-dependent component of mTOR-catalyzed p70S6k phosphorylation in vitro. Raptor appears to serve as an mTOR scaffold protein, the binding of which to the TOS motif of mTOR substrates is necessary for effective mTOR-catalyzed phosphorylation in vivo and perhaps for conferring S6K and 4E-BP1 sensitivity to rapamycin and amino acid sufficiency (Nojima, 2003).
The mammalian target of rapamycin, mTOR, is a serine/threonine kinase that controls cell growth and proliferation via the translation regulators eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1) and ribosomal protein S6 kinase 1 (S6K1). A TOR signaling (TOS) motif is present in the N terminus of S6K1 and the C terminus of 4E-BP1. In S6K1, the TOS motif is necessary to facilitate mTOR signaling to phosphorylate and activate S6K1. However, it has been unclear how the TOS motif in S6K1 and 4E-BP1 mediates mTOR signaling. A functional TOS motif is shown to be required for 4E-BP1 to bind to raptor (a recently identified mTOR-interacting protein), for 4E-BP1 to be efficiently phosphorylated in vitro by the mTOR/raptor complex, and for 4E-BP1 to be phosphorylated in vivo at all identified mTOR-regulated sites. mTOR/raptor-regulated phosphorylation is necessary for 4E-BP's efficient release from the translational initiation factor eIF4E. Consistently, overexpression of a mutant of 4E-BP1 containing a single amino acid change in the TOS motif (F114A) reduces cell size, demonstrating that mTOR-dependent regulation of cell growth by 4E-BP1 is dependent on a functional TOS motif. These data demonstrate that the TOS motif functions as a docking site for the mTOR/raptor complex, which is required for multisite phosphorylation of 4E-BP1, eIF4E release from 4E-BP1, and cell growth (Schalm, 2003).
The mammalian TOR (mTOR) pathway integrates nutrient- and growth factor-derived signals to regulate growth, the process whereby cells accumulate mass and increase in size. mTOR is a large protein kinase and the target of rapamycin, an immunosuppressant that also blocks vessel restenosis and has potential anticancer applications. mTOR interacts with the raptor and GΔL proteins to form a complex that is the target of rapamycin. mTOR is also part of a distinct complex defined by the novel protein rictor (rapamycin-insensitive companion of mTOR). Rictor shares homology with the previously described pianissimo from D. discoidieum, STE20p from S. pombe, and AVO3p from S. cerevisiae. Interestingly, AVO3p is part of a rapamycin-insensitive TOR complex that does not contain the yeast homolog of raptor and signals to the actin cytoskeleton through PKC1. Consistent with this finding, the rictor-containing mTOR complex contains GΔL but not raptor, and it neither regulates the mTOR effector S6K1 nor is it bound by FKBP12-rapamycin. The rictor-mTOR complex modulates the phosphorylation of Protein Kinase C alpha (PKCa) and the actin cytoskeleton, suggesting that this aspect of TOR signaling is conserved between yeast and mammals (Sarbassov, 2004).
Deregulation of Akt/protein kinase B (PKB) is implicated in the pathogenesis of cancer and diabetes. Akt/PKB activation requires the phosphorylation of Thr308 in the activation loop by the phosphoinositide-dependent kinase 1 (PDK1) and Ser473 within the carboxyl-terminal hydrophobic motif by an unknown kinase. This study shows that in Drosophila and human cells the target of rapamycin (TOR) kinase and its associated protein rictor are necessary for Ser473 phosphorylation and that a reduction in rictor or mammalian TOR (mTOR) expression inhibited an Akt/PKB effector. The rictor-mTOR complex directly phosphorylated Akt/PKB on Ser473 in vitro and facilitated Thr308 phosphorylation by PDK1. Rictor-mTOR may serve as a drug target in tumors that have lost the expression of PTEN, a tumor suppressor that opposes Akt/PKB activation (Sarbassov, 2005b).
The raptor-mTOR protein complex is a key component of a nutrient-sensitive signaling pathway that regulates cell size by controlling the accumulation of cellular mass. How nutrients regulate signaling through the raptor-mTOR complex is not well known. This study shows that a redox-sensitive mechanism regulates the phosphorylation of the raptor-mTOR effector S6K1, the interaction between raptor and mTOR, and the kinase activity of the raptor-mTOR complex. In cells treated with the oxidizing agents diamide or phenylarsine oxide, S6K1 phosphorylation increased and became insensitive to nutrient deprivation. Conversely, the reducing reagent BAL (British anti-Lewisite, also known as 2,3-dimercapto-1-propanol) inhibits S6K1 phosphorylation and stabilizes the interaction of mTOR and raptor to mimic the state of the complex under nutrient-deprived conditions. These findings suggest that a redox-based signaling mechanism may participate in regulating the nutrient-sensitive raptor-mTOR complex and pathway (Sarbassov, 2005b).
The drug rapamycin has important uses in oncology, cardiology, and transplantation medicine, but its clinically relevant molecular effects are not understood. When bound to FKBP12, rapamycin interacts with and inhibits the kinase activity of a multiprotein complex composed of mTOR, mLST8, and raptor (mTORC1). The distinct complex of mTOR, mLST8, and rictor (mTORC2) does not interact with FKBP12-rapamycin and is not thought to be rapamycin sensitive. mTORC2 phosphorylates and activates Akt/PKB, a key regulator of cell survival. This study shows that rapamycin inhibits the assembly of mTORC2 and that, in many cell types, prolonged rapamycin treatment reduces the levels of mTORC2 below those needed to maintain Akt/PKB signaling. The proapoptotic and antitumor effects of rapamycin are suppressed in cells expressing an Akt/PKB mutant that is rapamycin resistant. This work describes an unforeseen mechanism of action for rapamycin that suggests it can be used to inhibit Akt/PKB in certain cell types (Sarbassov, 2006b).
The mTOR complex 2 (mTORC2) containing mTOR and rictor is thought to be rapamycin-insensitive, and was recently shown to regulate the pro-survival kinase AKT by phosphorylation on Ser473. This study investigated the molecular effects of mTOR inhibition by the rapamycin derivatives (RDs) temsirolimus (CCI-779) and everolimus (RAD001) in AML cells. Unexpectedly, RDs not only inhibited the mTOR complex 1 (mTORC1) containing mTOR and raptor with decreased p70S6K, 4EPB1 phosphorylation and glut-1 mRNA, but also blocked AKT activation via inhibition of mTORC2 formation. This resulted in suppression of phosphorylation of the direct AKT substrate FKHR and decreased transcription of D-cyclins in AML cells. Similar observations were made in samples from patients with hematological malignancies who received RDs in clinical studies. This study provides first evidence that rapamycin derivatives inhibit AKT signaling in primary AML cells both in vitro and in vivo, and support the therapeutic potential of mTOR inhibition strategies in leukemias (Zeng, 2006).
While much is known about the molecular pathways that regulate embryonic development and adult homeostasis of the endocrine pancreas, little is known about what regulates early postnatal development and maturation of islets. Given that birth marks the first exposure to enteral nutrition, this study investigated how nutrient-regulated signaling pathways influence postnatal islet development. To do this loss-of-function studies were performed of mechanistic target of rapamycin (mTOR; see Drosophila Tor), a highly conserved kinase within a nutrient-sensing pathway known to regulate cellular growth, morphogenesis and metabolism. Deletion of mTOR in pancreatic endocrine cells had no significant effect on their embryonic development. However, within the first 2 weeks after birth, mTOR-deficient islets became dysmorphic, beta-cell maturation and function was impaired, and animals lost islet mass. Moreover, it was discovered that these distinct functions of mTOR are mediated by separate downstream branches of the pathway, in that mTORC1 (Raptor; see Drosophila Raptor) is the main complex mediating maturation and function of islets, whereas mTORC2 (Rictor; see Drosophila Rictor) impacts islet mass and architecture. Taken together, these findings suggest that nutrient-sensing may be a trigger for postnatal beta cell maturation and islet development (Sinagoga, 2017).
The enzyme mTOR (mammalian target of rapamycin) is a major target for therapeutic intervention to treat many human diseases, including cancer, but very little is known about the processes that control levels of mTOR protein. This study shows that mTOR is targeted for ubiquitination and consequent degradation by binding to the tumor suppressor protein FBXW7. Human breast cancer cell lines and primary tumors showed a reciprocal relation between loss of FBXW7 and deletion or mutation of PTEN (phosphatase and tensin homolog), which also activates mTOR. Tumor cell lines harboring deletions or mutations in FBXW7 are particularly sensitive to rapamycin treatment, which suggests that loss of FBXW7 may be a biomarker for human cancers susceptible to treatment with inhibitors of the mTOR pathway (Mao, 2008).
The mTOR kinase controls cell growth, proliferation, and survival through two distinct multiprotein complexes, mTORC1 and mTORC2. mTOR and mLST8 are in both complexes, while raptor and rictor are part of only mTORC1 and mTORC2, respectively. To investigate mTORC1 and mTORC2 function in vivo, mice deficient for raptor, rictor, or mLST8 were generated. Like mice null for mTOR, those lacking raptor die early in development. However, mLST8 null embryos survive until e10.5 and resemble embryos missing rictor. mLST8 is necessary to maintain the rictor-mTOR, but not the raptor-mTOR, interaction, and both mLST8 and rictor are required for the hydrophobic motif phosphorylation of Akt/PKB and PKCα, but not S6K1. Furthermore, insulin signaling to FOXO3, but not to TSC2 or GSK3β, requires mLST8 and rictor. Thus, mTORC1 function is essential in early development, mLST8 is required only for mTORC2 signaling, and mTORC2 is a necessary component of the Akt-FOXO and PKCα pathways (Guertin, 2007).
The mammalian target of rapamycin (mTOR) kinase is the catalytic subunit of at least two distinct signaling complexes, referred to as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Studies with cultured cell lines indicate that the complexes participate in different pathways and recognize distinct substrates, the specificity of which is determined by unique mTOR-interacting proteins. mTORC1 controls cell growth in part by phosphorylating S6 Kinase 1 (S6K1) and the eIF-4E-binding protein 1 (4E-BP1), known regulators of protein synthesis. It has been proposed that mTORC2 phosphorylates and activates Akt/PKB, which regulates cell proliferation, growth, survival, and metabolism. Full activation of Akt/PKB requires phosphorylation of S473 of the hydrophobic motif, the site proposed to depend on mTORC2, as well as phosphorylation of T308 of the activation loop by PDK1. The clinically valuable drug rapamycin specifically inhibits mTORC1 activity, although recent studies indicate that prolonged rapamycin treatment can also inhibit mTORC2 assembly and function in some cell types. Both complexes participate in signaling pathways associated with human diseases, including tuberous sclerosis complex (TSC), lymphangioleiomyomatosis (LAM), Cowden disease, Peutz-Jeghers syndrome (PJS), neurofibromatosis, familial cardiac hypertrophy, and cancers characterized by hyperactivation of PI3K/Akt (Guertin, 2007).
The roles of mTORC1 and mTORC2 during mammalian development are not well understood. In addition to mTOR, mTORC1 contains raptor (regulatory-associated protein of mTOR) and mLST8 (also called GβL). mTORC2 also contains mTOR and mLST8, but instead of raptor, this complex contains rictor (rapamycin-insensitive companion of mTOR). Germline disruption of mTOR in mice causes embryonic lethality at or around implantation. However, when these mice were engineered, it was not known that mTOR is part of two distinct complexes and pathways. With this new information, it becomes difficult to interpret the phenotypes of the mTOR null mice because these animals lack both mTORC1 and mTORC2 function. Thus, to determine the in vivo role of each branch of the mTOR signaling network, mice lacking the expression of raptor, rictor, and mLST8 were generated and characterized. It is conclude that during mammalian development, mTORC1 and mTORC2 have different, but essential, roles; that mLST8 is required only for mTORC2 function; and that mTORC2 is a crucial component of the Akt/PKB-FOXO and PKCα signaling networks (Guertin, 2007).
Much of the mammalian skeleton is derived from a cartilage template that undergoes rapid growth during embryogenesis, but the molecular mechanism of growth regulation is not well understood. Signaling by mammalian target of rapamycin complex 1 (mTORC1) is an evolutionarily conserved mechanism that controls cellular growth. This study reports that mTORC1 signaling is activated during limb cartilage development in the mouse embryo. Disruption of mTORC1 signaling through deletion of either mTOR or the associated protein Raptor greatly diminishes embryonic skeletal growth associated with severe delays in chondrocyte hypertrophy and bone formation. The growth reduction of cartilage is not due to changes in chondrocyte proliferation or survival, but is caused by a reduction in cell size and in the amount of cartilage matrix. Metabolic labeling reveals a notable deficit in the rate of protein synthesis in Raptor-deficient chondrocytes. Thus, mTORC1 signaling controls limb skeletal growth through stimulation of protein synthesis in chondrocytes (Chen, 2014).
Many forms of long-lasting behavioral and synaptic plasticity require the synthesis of new proteins. For example, long-term potentiation (LTP) that endures for more than an hour requires both transcription and translation. The signal-transduction mechanisms that couple synaptic events to protein translational machinery during long-lasting synaptic plasticity, however, are not well understood. One signaling pathway that is stimulated by growth factors and results in the translation of specific mRNAs includes the rapamycin-sensitive kinase mammalian target of rapamycin (mTOR, also known as FRAP and RAFT-1). Several components of this translational signaling pathway, including mTOR, eukaryotic initiation factor-4E-binding proteins 1 and 2, and eukaryotic initiation factor-4E, are present in the rat hippocampus as shown by Western blot analysis, and these proteins are detected in the cell bodies and dendrites in the hippocampal slices by immunostaining studies. In cultured hippocampal neurons, these proteins are present in dendrites and are often found near the presynaptic protein, synapsin I. At synaptic sites, their distribution completely overlaps with a postsynaptic protein, PSD-95. These observations suggest the postsynaptic localization of these proteins. Disruption of mTOR signaling by rapamycin results in a reduction of late-phase LTP expression induced by high-frequency stimulation; the early phase of LTP is unaffected. Rapamycin also blocks the synaptic potentiation induced by brain-derived neurotrophic factor in hippocampal slices. These results demonstrate an essential role for rapamycin-sensitive signaling in the expression of two forms of synaptic plasticity that require new protein synthesis. The localization of this translational signaling pathway at postsynaptic sites may provide a mechanism that controls local protein synthesis at potentiated synapses (Tang, 2002).
Balanced control of neural progenitor maintenance and neuron production is crucial in establishing functional neural circuits during brain development, and abnormalities in this process are implicated in many neurological diseases. However, the regulatory mechanisms of neural progenitor homeostasis remain poorly understood. This study shows that mammalian target of rapamycin (mTOR) is required for maintaining neural progenitor pools and plays a key role in mediating glycogen synthase kinase 3 (GSK3; see Drosophila Shaggy) signaling during brain development. First, conditional mutant mice exhibiting deletion of mTOR were generated and characterized in neural progenitors and neurons in the developing brain using Nestin-cre and Nex-cre lines, respectively. The elimination of mTOR resulted in abnormal cell cycle progression of neural progenitors in the developing brain and thereby disruption of progenitor self-renewal. Accordingly, production of intermediate progenitors and postmitotic neurons were markedly suppressed. Next, it was discovered that GSK3, a master regulator of neural progenitors, interacts with mTOR and controls its activity in cortical progenitors. Finally, it was found that inactivation of mTOR activity suppresses the abnormal proliferation of neural progenitors induced by GSK3 deletion. These findings reveal that the interaction between mTOR and GSK3 signaling plays an essential role in dynamic homeostasis of neural progenitors during brain development (Ka, 2014).
Mammalian target of rapamycin (mTOR) integrates multiple signals, including nutrient status, growth factor availability, and stress, to regulate cellular and organismal growth. How mTOR regulates transcriptional programs in response to these diverse stimuli is poorly understood. MondoA and its obligate transcription partner Mlx are basic helix-loop-helix leucine zipper (bHLHZip) transcription factors that sense and execute a glucose-responsive transcriptional program. MondoA-Mlx complexes (See Drosophila Mondo and Bigmax) activate expression of thioredoxin-interacting protein (TXNIP), which is a potent inhibitor of cellular glucose uptake and aerobic glycolysis. Both mTOR and MondoA are central regulators of glucose metabolism, yet whether they interact physically or functionally is unknown. This study shows that inhibition of mTOR induces MondoA-dependent expression of TXNIP, coinciding with reduced glucose uptake. Mechanistically, mTOR binds to MondoA in the cytoplasm and prevents MondoA-Mlx complex formation, restricting MondoA's nuclear entry and reducing TXNIP expression. Further, This study shows that mTOR inhibitors and reactive oxygen species (ROS) regulate interaction between MondoA and mTOR in an opposing manner. Like mTOR's suppression of the MondoA-TXNIP axis, MondoA can also suppress mTOR complex 1 (mTORC1) activity via its direct transcriptional regulation of TXNIP. Collectively, these studies reveal a regulatory relationship between mTOR and the MondoA-TXNIP axis that is proposed to contributes to glucose homeostasis (Kaadige, 2015).
Dendritic arbors are compartments of neurons dedicated to receiving synaptic inputs. Their shape is an outcome of both the intrinsic genetic program and environmental signals. The microtubules and actin cytoskeleton are both crucial for proper dendritic morphology, but how they interact is unclear. The present study demonstrates that microtubule plus-end tracking protein CLIP-170 (ortholog of Drosophila CLIP-190) and actin-binding protein IQGAP1 regulate dendrite morphology of rat neurons by coordinating the interaction between microtubules and the actin cytoskeleton. Moreover, this study shows that mTOR kinase (see Drosophila Tor) interacts with CLIP-170 and is needed for efficient formation of a protein complex containing CLIP-170 and IQGAP1. Dynamic microtubules, CLIP-170, and IQGAP1 are required for proper dendritic arbor morphology and PI3K-mTOR-induced increase in dendritic arbor complexity. Moreover, CLIP-170 and IQGAP1 knockdown modulates dendritic arbor growth via regulation of the actin cytoskeleton. It is postulated that mTOR controls dendritic arbor morphology by enhancing cross talk between dynamic microtubules and actin through CLIP-170 and IQGAP1 (Swiech, 2011).
Dendrites are the major receivers of synaptic inputs in the nervous system. Their development involves several cellular processes, among which both cytoskeletal rearrangement and stabilization play important roles. Microtubules are extremely dynamic structures involved in transport of cellular cargo, maintenance of cell shape, and regulation of cell motility. The dynamic behavior of microtubules is crucial for their function and occurs primarily at the microtubule plus-ends under the control of a heterogeneous family of plus-end tracking proteins (+TIPs). In addition to direct regulation of microtubule dynamics, +TIPs are important for intracellular transport and cross talk between microtubules and the actin cytoskeleton. In neurons, +TIPs were shown to regulate neuronal survival, migration, and polarization, axon navigation, spine growth and maintenance, and synaptic plasticity. However, the role of +TIPs in dendritic arborization has not yet been studied extensively (Swiech, 2011).
Cytoplasmic linker protein of 170 kDa (CLIP-170), a founding member of the +TIP family, is a multifunctional protein whose actions are defined by protein partners interacting on its N and C termini. Interactions between CLIP-170 and the IQ motif-containing GTPase-activating protein 1 (IQGAP1) provide a possible link between microtubules and the actin cytoskeleton by bridging microtubule plus-ends to the cell cortex. Notably, +TIP-dependent cross talk between dynamic microtubules and actin is crucial for proper axonal growth and spine maintenance but has not been studied in the context of dendritic arborization (Swiech, 2011).
CLIP-170 has been shown to be phosphorylated at multiple sites in vivo, and its phosphorylation is sensitive to rapamycin, an mTOR kinase inhibitor. Intriguingly, mTOR activity was previously shown to be indispensable for proper dendritic arbor development in hippocampal neurons. Altogether, these observations suggest that CLIP-170 may be one of the important downstream effectors of mTOR kinase for neuronal morphology and raise the question of how mTOR regulates CLIP-170 activity as needed for proper dendritic arborization (Swiech, 2011).
Thid pstudy revealed that dynamic microtubules and CLIP-170 are needed for proper dendritic arbor morphology. Furthermore, both are required for class I phosphoinositide 3-kinase (PI3K)-induced, mTOR-dependent dendritic growth. Evidence that mTOR binds CLIP-170 and regulates its interaction with IQGAP1. Consequently, knockdown of IQGAP1 results in simplification of dendritic arborization and inhibition of PI3K-driven growth. Finally, this study shows that deterioration of dendritic morphology attributable to depletion of either CLIP-170 or IQGAP1 can be prevented by actin stabilization. Thus, these observations strongly suggest a need for mTOR coordinated interaction of dynamic microtubules with the actin cytoskeleton for proper dendritic arbor morphology (Swiech, 2011).
This study describes a novel mechanism of control of dendritic arbor morphology in mammalian neurons via a +TIP family member-CLIP-170. This mechanism likely involves an mTOR-dependent interaction between CLIP-170 and IQGAP1, an actin-regulating protein that coordinates cross talk between dynamic microtubule plus-ends and the actin cytoskeleton. Interactions of CLIP-170 with IQGAP1 and subsequently F-actin are decreased by mTOR inhibition in neuronal cells. Both CLIP-170 and IQGAP1, similarly to mTOR, are indispensible for proper dendritic arbor morphology, and dendritic arbor deterioration caused by their knockdown can be rescued by F-actin stabilization (jasplakinolide treatment). Moreover, the presence of CLIP-170 and IQGAP1 and microtubule dynamics are needed to support mTOR-dependent, PI3K-induced increases in dendritic arbor complexity in rat hippocampal neurons. These observations suggest that in addition to previously described mTOR-dependent translational control of dendritogenesis, this kinase supports this process by enhancing interactions between microtubule and actin-binding proteins. This, in turn, can lead to anchoring and stabilization of microtubules or to microtubule-induced changes in actin dynamics needed for dendritic growth or stabilization (Swiech, 2011).
In different model organisms, proper dendritic arbor morphology depends on proteins to ensure proper regulation of microtubule polymerization (CRMP3, Cypin), stabilization (MAP1, MAP2, doublecortin), severing (spastin), and depolymerization (SCLIP). These observations strongly suggest the importance of microtubule dynamics for dendritic tree morphology. Therefore, it is surprising how little is known about the role of +TIPs that are crucial for microtubule dynamics during dendritic arborization. This is in contrast to numerous reports describing the contribution of +TIPs to neuronal polarization or axonal growth Additionally, the importance of +TIPs for dendritic spine morphology was recently proved. The present study provides evidence that knockdown of CLIP-170, a founding member of the +TIP protein family, is vital for several aspects of dendritic arbor morphology (Swiech, 2011).
Ample evidence supporting the conclusion that +TIPs are important for dendritic growth derives from studies in Drosophila. At least three proteins, DLIS-1, Dhc64, and shortstop (also known as kakapo), respective homologs of Lis1, dynein heavy chain, and ACF7, were shown to regulate dendritic arborization in fruit flies. The question remains, however, how +TIPs contribute to dendritic growth. It has been postulated that dynein-dependent transport of 'branching machinery' from the cell soma toward distal dendrite regions is crucial for proper patterning of dendritic arborization (da) neurons. Indeed, in proximal dendrites of Drosophila and in mammalian neurons, the majority of microtubules have their plus-ends oriented toward the cell soma, and dynein transport is a major route of cargo sorting into dendrites. The identity of this machinery remains largely unknown, but Rab5 endosomes have emerged as a component. +TIPs can also contribute to dendrite growth in other ways. All three +TIPs (Lis1, dynein, and ACF7) can directly or indirectly link microtubules to the cell cortex or actin cytoskeleton, another important factor for dendritic patterning. Whether these particular proteins can regulate dendritic arborization by such cross-linking remains to be established, but the work presented herein strongly supports such a scenario for CLIP-170 (Swiech, 2011).
In principle, each of the proposed functions for CLIP-170 (i.e., regulation of microtubule dynamics, facilitation of dynein-dependent transport, and cross-linking microtubules to the actin cytoskeleton) may importantly contribute to CLIP-170's effects on proper dendritic morphology. However, neither N- nor C-terminal deletion mutants could fully rescue the CLIP-170 knockdown phenotype. Protein interactions of the N terminus supporting net microtubule growth could potentially allow the advancement of microtubules into a newly formed filopodium and help branch formation. CLIP-170 also interacts via its C terminus with p150Glued, a part of the dynein-dynactin complex, and Lis1, a role for which in dendrites was discussed above. The present study focused on cross-linking two types of cytoskeletons. Interactions between +TIPs and actin-regulating proteins have been shown to be a basis for morphological changes in neurons, including the formation of neurites (EB3 and drebrin) and mature dendritic spines (EB3 and p140Cap). This study reports that CLIP-170 can interact with IQGAP1, and both proteins regulate dendritic arborization in an actin-dependent fashion (Swiech, 2011).
IQGAP1 has been shown to transiently stabilize microtubules at the cell cortex of non-neuronal cells by interaction with CLIP-170. Based on these observations, CLIP-170 and IQGAP1 may coordinate dendritic growth simply by stabilizing microtubules at a site of a new dendritic branch formation. This would transiently polarize and enhance transport along microtubules toward future dendrites. A transport-only mechanism does not explain, however, why jasplakinolide treatment prevents dendritic arbor deterioration caused by either CLIP-170 or IQGAP1 knockdown. This observation suggests that in addition to microtubule stabilization at the cell cortex, CLIP-170-IQGAP1-dependent dendritic growth also involves interdependent changes in actin cytoskeleton. Indeed, the CLIP-170 interaction with IQGAP1 can also take part in actin regulation. First, in vitro IQGAP1 stimulates actin polymerization as well as actin cross-linking. Second, it has been shown that in granular cerebellar and neonatal hippocampal neurons, CLIP-170 and IQGAP1 are in a protein complex that regulates F-actin at a leading edge of migrating neurons, in growing neurites, and somal plasma membrane. In addition, this work shows that (1) CLIP-170 requires IQGAP1 and mTOR to bind to F-actin and (2) negative effects of CLIP-170 and IQGAP1 knockdown on dendritic arborization are interdependent and can be rescued by actin stabilization, forced by jasplakinolide. Taking all the above into consideration, although alternative scenarios cannot be ruled out, in which CLIP-170 and IQGAP1 may regulate F-actin independent of each other, it is postulated that formation of a CLIP-170-IQGAP1 complex could be involved in shaping dendritic arbors by mTOR, transiently stabilizing and cross-linking microtubules with the cellular cortex, and regulating F-actin (Swiech, 2011).
In previous work, it was shown that mTOR-dependent dendritic arborization requires protein synthesis. mTOR kinase is best known for its key role in the control of translation, and axon formation, for example, has been shown to be regulated by mTOR-driven translation of microtubule-regulating proteins such as SAD kinases or CRMP-2. Although newly translated proteins important for mTOR-dependent dendritic growth have not yet been identified, several candidates are among those proteins locally translated in dendrites in response to brain-derived neurotrophic factor (BDNF) or insulin. However, the present study shows that, in addition to protein translation, PI3K-driven dendritic arborization, which is an mTOR-controlled process, depends on microtubule dynamics and the presence of both CLIP-170 and IQGAP1. Moreover, this study confirms that CLIP-170 is an mTOR kinase substrate and reveal mTOR-dependent regulation of CLIP-170 protein-protein interactions (Swiech, 2011).
PI3K activates mTORC1 in response to several stimuli known to induce dendritic growth, such as neurotransmitters (e.g., glutamate), neurotrophins (e.g., BDNF), and the extracellular matrix (e.g., chicken acidic leucine-rich epidermal growth factor-like domain-containing brain protein/neuroglycan C). However, active PI3K also induces small GTPases such as Rac1 and Cdc42 that interact with IQGAP1. Thus, it is speculated that activation of mTOR by extracellular signals may allow the formation of the IQGAP1-CLIP-170 complex with PI3K-activated Cdc42 or Rac1 to converge mTOR and small Rho GTPase signaling pathways downstream of PI3K to coordinate the regulation of microtubules and actin during dendritic growth. The present study adds the control of microtubule-actin interactions to the repertoire of mTOR during dendritic arbor growth (Swiech, 2011).
The poor axon regeneration in the central nervous system (CNS) often leads to permanent functional deficit following disease or injury. For example, degeneration of retinal ganglion cell (RGC) axons in glaucoma leads to irreversible loss of vision. This study tested the hypothesis that the mTOR pathway regulates the development of human RGCs and that its recruitment after injury facilitates axon regeneration. The mTOR pathway is active during RGC differentiation, and using the induced pluripotent stem cell model of neurogenesis this study shows that it facilitates the differentiation, function and neuritogenesis of human RGCs. Using a microfluidic model, it was demonstrated that recruitment of the mTOR pathway facilitates human RGC axon regeneration after axotomy, providing evidence that the recapitulation of developmental mechanism(s) might be a viable approach for facilitating axon regeneration in the diseased or injured human CNS, thus helping to reduce and/or recover loss of function (Teotia, 2019).
Mammalian target of rapamycin (mTOR: See Drosophila Tor) is a highly conserved eukaryotic protein kinase that coordinates cell growth and metabolism, and plays a critical role in cancer, immunity, and aging. It remains unclear how mTOR signaling in individual tissues contributes to whole-organism processes because mTOR inhibitors, like the natural product rapamycin, are administered systemically and target multiple tissues simultaneously. This study developed a chemical-genetic system, termed selecTOR, that restricts the activity of a rapamycin analog to specific cell populations through targeted expression of a mutant FKBP12 protein. This analog has reduced affinity for its obligate binding partner FKBP12, which reduces its ability to inhibit mTOR in wild-type cells and tissues. Expression of the mutant FKBP12, which contains an expanded binding pocket, rescues the activity of this rapamycin analog. Using this system, it was shown that selective mTOR inhibition can be achieved in Saccharomyces cerevisiae and human cells, and The utility of this system was developed in an intact metazoan model organism the tissues responsible for a rapamycin-induced developmental delay in Drosophila were identified (Wassarman, 2022).
Search PubMed for articles about Drosophila Target of rapamycin
Alarcon, C. M., Heitman, J. and Cardenas, M. E. (1999). Protein kinase activity and identification of a toxic effector domain of the target of rapamycin TOR proteins in yeast. Mol. Biol. Cell 10: 2531-2546. 10436010
Alic, N., Andrews, T. D., Giannakou, M. E., Papatheodorou, I., Slack, C., Hoddinott, M. P., Cocheme, H. M., Schuster, E. F., Thornton, J. M. and Partridge, L. (2011). Genome-wide dFOXO targets and topology of the transcriptomic response to stress and insulin signalling. Mol Syst Biol 7: 502. PubMed ID: 21694719
Arimbasseri, A. G. and Maraia, R. J. (2016). RNA polymerase III advances: structural and tRNA functional views. Trends Biochem. Sci. 41: 546-559. PubMed ID: 27068803
Armstrong, A. R., Laws, K. M. and Drummond-Barbosa, D. (2014). Adipocyte amino acid sensing controls adult germline stem cell number via the amino acid response pathway and independently of Target of Rapamycin signaling in Drosophila. Development 141: 4479-4488. PubMed ID: 25359724
Avet-Rochex, A., Carvajal, N., Christoforou, C. P., Yeung, K., Maierbrugger, K. T., Hobbs, C., Lalli, G., Cagin, U., Plachot, C., McNeill, H., Bateman, J. M. (2014). Unkempt is negatively regulated by mTOR and uncouples neuronal differentiation from growth control. PLoS Genet 10: e1004624. PubMed ID: 25210733
Bai, X., et al. (2007). Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 318(5852): 977-80. PubMed citation: 17991864
Barbet, N. C., Schneider, U., Helliwell, S. B., Stansfield, I., Tuite, M. F. and Hall, M. N. (1996). TOR controls translation initiation and early G1 progression in yeast. Mol. Biol. Cell 7: 25-42. PubMed Citation: 8741837
Bar-Peled, L., Chantranupong, L., Cherniack, A. D., Chen, W. W., Ottina, K. A., Grabiner, B. C., Spear, E. D., Carter, S. L., Meyerson, M. and Sabatini, D. M. (2013). A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340: 1100-1106. PubMed ID: 23723238
Beck, T. and Hall, M. N. (1999). The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402: 689-692. PubMed Citation: 10604478
Berry, D. L., and Baehrecke, E. H. (2007). Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 131: 1137-1148. PubMed Citation: 18083103
Beugnet, A., Wang, X. and Proud, C. G. (2003). The TOR-signaling and RAIP motifs play distinct roles in the mTOR-dependent phosphorylation of initiation factor 4E-binding protein 1 in vivo. J. Biol. Chem. 12912989
Brown, E. J., Albers, M. W., Shin, T. B., Ichikawa, K., Keith, C. T., Lane, W. S., and Schreiber, S. L. (1994). A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369: 756-758. 8008069
Bryk, B., Hahn, K., Cohen, S. M. and Teleman, A. A. (2010). MAP4K3 regulates body size and metabolism in Drosophila. Dev. Biol. 344(1): 150-7. PubMed Citation: 20457147
Boehlke, C., et al. (2010). Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 12(11): 1115-22. PubMed Citation: 20972424
Brugarolas, J., et al. (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004 18(23): 2893-904. 15545625
Bryk, B., Hahn, K., Cohen, S. M. and Teleman, A. A. (2010). MAP4K3 regulates body size and metabolism in Drosophila. Dev. Biol. 344(1): 150-7. PubMed Citation: 20457147
Budanov, A. V. and Karin, M. (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134: 451-60. PubMed Citation: 18692468
Cai, W., Wei, Y., Jarnik, M., Reich, J. and Lilly, M. A. (2016). The GATOR2 component Wdr24 regulates TORC1 activity and lysosome function. PLoS Genet 12: e1006036. PubMed ID: 27166823
Cardenas, M. E., et al. (1999). The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 13: 3271-3279. PubMed Citation: 10617575
Chang, Y.Y. amd Neufeld, T. P. (2009). An Atg1/Atg13 complex with multiple roles in tor-mediated autophagy regulation. Mol. Biol. Cell. 20(7): 2004-14. PubMed Citation: 19225150
Chell, J. M. and Brand, A. H. (2010). Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell 143: 1161-1173. PubMed Citation: 21183078
Chen, C. C., et al. (2010). FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev. Cell 18(4): 592-604. PubMed Citation: 20412774
Chen, J. and Long, F. (2014). mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis. Development 141: 2848-2854. PubMed ID: 24948603
Choi, J. H., et al. (2000). TOR signaling regulates microtubule structure and function. Curr. Biol. 10: 861-864. PubMed Citation: 10899009
Choi, K. M., McMahon, L. P. and Lawrence, J. C. (2003). Two motifs in the translational repressor PHAS-I required for efficient phosphorylation by mammalian target of rapamycin and for recognition by raptor. J. Biol. Chem. 278(22): 19667-73. 12665511
Chou, M.M. and Blenis, J. 1995. The 70 kDa S6 kinase: Regulation of a kinase with multiple roles in mitogenic signaling. Curr. Opin. Cell Biol. 7: 806-814. 8608011
Coelho, C. M. and Leevers, S. J. (2000}. Do growth and cell division rates determine cell size in multicellular organisms? J. Cell Sci. 113: 2927-2934. 10934032
Colombani, J., et al. (2003). A nutrient sensor mechanism controls Drosophila growth. Cell 114: 739-749. Medline abstract: 14505573
Cutler, N. S., Heitman, J. and Cardenas, M. E. (1999). TOR kinase homologs function in a signal transduction pathway that is conserved from yeast to mammals. Mol. Cell Endocrinol. 155: 135-142. 10580846
Cutler, N. S., Pan, X., Heitman, J. and Cardenas, M. E. (2001). The TOR signal transduction cascade controls cellular differentiation in response to nutrients. Mol. Biol. Cell 12(12): 4103-13. 11739804
Dan, H. C., et al. (2008). Akt-dependent regulation of NF-kappaB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 22(11): 1490-500. PubMed Citation: 18519641
Dennis, P. B., Fumagalli, S. and Thomas, G. (1999). Target of rapamycin (TOR): Balancing the opposing forces of protein synthesis and degradation. Curr. Opin. Genet. Dev. 9: 49-54. 10072357
Demetriades, C., Doumpas, N. and Teleman, A. A. (2014). Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156: 786-799. PubMed ID: 24529380
Dokudovskaya, S., Waharte, F., Schlessinger, A., Pieper, U., Devos, D. P., Cristea, I. M., Williams, R., Salamero, J., Chait, B. T., Sali, A., Field, M. C., Rout, M. P. and Dargemont, C. (2011). A conserved coatomer-related complex containing Sec13 and Seh1 dynamically associates with the vacuole in Saccharomyces cerevisiae. Mol Cell Proteomics 10: M110 006478. PubMed ID: 21454883
Dutta, S. and Baehrecke, E. H. (2008). Warts is required for PI3K-regulated growth arrest, autophagy, and autophagic cell death in Drosophila. Curr. Biol. 18(19): 1466-75. PubMed Citation: 18818081
Filer, D., Thompson, M. A., Takhaveev, V., Dobson, A. J., Kotronaki, I., Green, J. W. M., Heinemann, M., Tullet, J. M. A. and Alic, N. (2017). RNA polymerase III limits longevity downstream of TORC1. Nature 552(7684): 263-267. PubMed ID: 29186112
Findlay, G. M., Yan, L., Procter, J., Mieulet, V. and Lamb, R. F. (2007). A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem. J. 403(1): 13-20. PubMed Citation: 17253963
Frias, M. A., Mukhopadhyay, S., Lehman, E., Walasek, A., Utter, M., Menon, D. and Foster, D. A. (2019). Phosphatidic acid drives mTORC lysosomal translocation in the absence of amino acids. J Biol Chem 295(1):263-274. PubMed ID: 31767684
Foerster, P., Daclin, M., Asm, S., Faucourt, M., Boletta, A., Genovesio, A. and Spassky, N. (2017). mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development 144(2): 201-210. PubMed ID: 27993979
Gancz, D. and Gilboa, L. (2013). Insulin and Target of rapamycin signaling orchestrate the development of ovarian niche-stem cell units in Drosophila. Development 140: 4145-4154. PubMed ID: 24026119
Gao, X., et al. (2002). Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat. Cell Biol. 4(9): 699-704. 12172555
Giordano, E., Peluso, I., Senger, S. and Furia, M. (1999). minifly, a Drosophila gene required for ribosome biogenesis. J. Cell Biol. 144: 1123-1133. Medline abstract: 10087258
Grandison, R. C., Piper, M. D. and Partridge, L. (2009). Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 462: 1061-1064. PubMed Citation: 19956092
Gomez-Roman, N., Grandori, C., Eisenman, R. N. and White, R. J. (2003). Direct activation of RNA polymerase III transcription by c-Myc. Nature 421(6920): 290-4. PubMed Citation: 12529648
Grewal, S. S., Evans, J. R. and Edgar, B. A (2007). Drosophila TIF-IA is required for ribosome synthesis and cell growth and is regulated by the TOR pathway. J. Cell Biol. 179: 1105-1113. PubMed Citation: 18086911
Grewal, S. S. (2009). Insulin/TOR signaling in growth and homeostasis: a view from the fly world. Int J Biochem Cell Biol 41: 1006-1010. PubMed Citation: 18992839
Guertin, D. A., et al. (2006). Functional genomics identifies TOR-regulated genes that control growth and division. Curr. Biol. 16(10): 958-70. Medline abstract: 16713952
Guertin, D. A., et al. (2007). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCa, but not S6K1. Dev. Cell. 11: 859-871. Medline abstract: 17141160
Hallsson, J. H., Haflidadöttir, B. S., Stivers, C., Odenwald, W., Arnheiter, H., Pignoni, F. and Steingrïmsson, E. (2004). The basic helix-loop-helix leucine zipper transcription factor Mitf is conserved in Drosophila and functions in eye development. Genetics 167: 233-241. PubMed ID: 15166150
Hara, K., et al. (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110(2): 177-89. 12150926
Hardwick, J. S., et al. (1999). Rapamycin-modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proc. Natl. Acad. Sci. 96(26): 14866-70. PubMed Citation: 10611304
Hari, K. L., et al. (1995). The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell 82: 815-821. PubMed Citation: 7671309
Heitman, J., Movva, N. R. and Hall, M. N. (1991). Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 253: 905-909. 1715094
Hennig, K.M. and Neufeld, T.P. (2002). Inhibition of cellular growth and proliferation by dTOR overexpression in Drosophila. Genesis 34: 107-110. 12324961
Hennig, K. M., Colombani, J. and Neufeld, T. P. (2006). TOR coordinates bulk and targeted endocytosis in the Drosophila melanogaster fat body to regulate cell growth. J. Cell Biol. 173(6): 963-74. Medline abstract: 16785324
Hermle, T., Guida, M. C., Beck, S., Helmstädter, S. and Simons, M. (2013). Drosophila ATP6AP2/VhaPRR functions both as a novel planar cell polarity core protein and a regulator of endosomal trafficking. EMBO J. 32: 245-259. PubMed ID: 23292348
Hiesinger, P. R., Fayyazuddin, A., Mehta, S. Q., Rosenmund, T., Schulze, K. L., Zhai, R. G., Verstreken, P., Cao, Y., Zhou, Y., Kunz, J. et al. (2005). The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell 121: 607-620. PubMed ID: 15907473
Hietakangas, V. and Cohen, S. M. (2007). Re-evaluating AKT regulation: role of TOR complex 2 in tissue growth. Genes Dev 21: 632-637. PubMed ID: 17369395
Huang, S., et al. (2003). Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21Cip1. Mol. Cell 11: 1491-1501. 12820963
Inoki, K., Li, Y., Zhu, T., Wu, J. and Guan, K. L. (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4(9): 648-57. 12172553
Isotani, S., et al. (2000). Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J. Biol. Chem. 274(48): 34493-8. PubMed Citation: 10567431
Jevtov, I., et al. (2015). TORC2 mediates the heat stress response in Drosophila by promoting the formation of stress granules. J Cell Sci 128(14): 2497-508. PubMed ID: 26054799
Jia, K. Chen, D. and Riddle, D. L. (2004). The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131: 3897-3906. 15253933
Juhasz, G., et al. (2008). The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J. Cell Biol. 181: 655-666. PubMed Citation: 18474623
Ka, M., Condorelli, G., Woodgett, J. R. and Kim, W. Y. (2014). mTOR regulates brain morphogenesis by mediating GSK3 signaling. Development 141: 4076-4086. PubMed ID: 25273085
Kaadige, M. R., Yang, J., Wilde, B. R. and Ayer, D. E. (2015). MondoA-Mlx transcriptional activity is limited by mTOR-MondoA interaction. Mol Cell Biol 35: 101-110. PubMed ID: 25332233
Kaeberlein, M., et al. (2005). Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310(5751): 1193-6. Medline abstract: 16293764
Kaeberlein, M., Powers, R. W., 3rd, Steffen, K. K., Westman, E. A., Hu, D., Dang, N., Kerr, E. O., Kirkland, K. T., Fields, S. and Kennedy, B. K. (2005). Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310: 1193-1196. PubMed ID: 16293764
Kamada, Y., Funakoshi, T., Shintani, T., Nagano, K., Ohsumi, M. and Ohsumi, Y. (2000). Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 150: 1507-1513. 10995454
Kapahi, K., et al. (2004). Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 14: 885-890. 15186745
Kauwe, G., Tsurudome, K., Penney, J., Mori, M., Gray, L., Calderon, M. R., Elazouzzi, F., Chicoine, N., Sonenberg, N. and Haghighi, A. P. (2016). Acute fasting regulates retrograde synaptic enhancement through a 4E-BP-dependent mechanism. Neuron 92(6): 1204-1212. PubMed ID: 27916456
Khurana, V., et al. (2006). TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 16: 230-241. 16461276
Kim, D. H., et al. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110(2): 163-75. 12150925
Kim, D. H., et al. (2003). GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 11(4): 895-904. 12718876
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P. and Guan, K. L. (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10: 935-945. PubMed ID: 18604198
Kim, W., Jang, Y. G., Yang, J. and Chung, J. (2017). Spatial activation of TORC1 is regulated by Hedgehog and E2F1 signaling in the Drosophila eye. Dev Cell 42(4): 363-375. PubMed ID: 28829944
Kim, Y. C., Park, H. W., Sciarretta, S., Mo, J. S., Jewell, J. L., Russell, R. C., Wu, X., Sadoshima, J. and Guan, K. L. (2014). Rag GTPases are cardioprotective by regulating lysosomal function. Nat Commun 5: 4241. PubMed ID: 24980141
Kirisako, T., Baba, M., Ishihara, N., Miyazawa, K., Ohsumi, M., Yoshimori, T., Noda, T. and Ohsumi, Y. (1999). Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 147: 435-446. 10525546
Koike-Kumagai, M., Yasunaga, K., Morikawa, R., Kanamori, T. and Emoto, K. (2009). The target of rapamycin complex 2 controls dendritic tiling of Drosophila sensory neurons through the Tricornered kinase signalling pathway. EMBO J 28: 3879-3892. PubMed ID: 19875983
Kumar, V., et al. (2000a). Functional interaction between RAFT1/FRAP/mTOR and protein kinase cdelta in the regulation of cap-dependent initiation of translation. EMBO J. 19: 1087-1097. PubMed Citation: 10698949
Kumar, V., et al. (2000b). Regulation of the rapamycin and FKBP-target 1/mammalian target of rapamycin and cap-dependent initiation of translation by the c-Abl protein-tyrosine kinase. J. Biol. Chem. 275(15): 10779-87. PubMed Citation: 10753870
Kuo, Y., Huang, H., Cai, T. and Wang, T. (2015). Target of Rapamycin Complex 2 regulates cell growth via Myc in Drosophila. Sci Rep 5: 10339. PubMed ID: 25999153
LaFever, L., Feoktistov, A., Hsu, H. J. and Drummond-Barbosa, D. (2010). Specific roles of Target of rapamycin in the control of stem cells and their progeny in the Drosophila ovary. Development 137(13): 2117-26. PubMed Citation: 20504961
Lawrence, J. C. and Abraham, R. T. (1997). PHAS/4E-BPs as regulators of mRNA translation and cell proliferation. Trends Biochem. Sci. 22: 345-349. 9301335
Lee, J. H., et al. (2010). Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 327: 1223-1228. PubMed Citation: 20203043
Luo, J., Liu, Y. and Nassel, D. R. (2013). Insulin/IGF-Regulated Size Scaling of Neuroendocrine Cells Expressing the bHLH Transcription Factor Dimmed in Drosophila. PLoS Genet 9: e1004052. PubMed ID: 24385933
Luong, N., et al. (2006). Activated FOXO-mediated insulin resistance is blocked by reduction of TOR activity. Cell Metab. 4(2): 133-42. Medline abstract: 16890541
Layalle, S., Arquier, N., Léopold, P. (2008). The TOR pathway couples nutrition and developmental timing in Drosophila. Dev. Cell 15(4): 568-77. PubMed Citation: 18854141
Mao, J. H., et al. (2008). FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321(5895): 1499-502. PubMed Citation: 18787170
Marshall L. (2008). Elevated RNA polymerase III transcription drives proliferation and oncogenic transformation. Cell Cycle 7(21): 3327-9. PubMed Citation: 18971635
Marshall, L., Rideout, E. J. and Grewal, S. S. (2012). Nutrient/TOR-dependent regulation of RNA polymerase III controls tissue and organismal growth in Drosophila. EMBO J. [Epub ahead of print]. PubMed Citation: 22367393
Marshansky, V., Rubinstein, J. L. and Grüber, G.(2014). Eukaryotic V-ATPase: novel structural findings and finctional insights. Biochim. Biophys. Acta. 1837: 857-879. PubMed ID: 24508215
Martin, D. E., Soulard, A. and Hall, M. N. (2004). TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell 119: 969-979. 15620355
Martin, D. N., Balgley, B., Dutta, S., Chen, J., Rudnick, P., Cranford, J., Kantartzis, S., DeVoe, D. L., Lee, C. and Baehrecke, E. H. (2007). Proteomic analysis of steroid-triggered autophagic programmed cell death during Drosophila development. Cell Death Differ. 14: 916-923. PubMed Citation: 17256009
Matsuo, T., Kubo, Y., Watanabe, Y. and Yamamoto, M. (2003). Schizosaccharomyces pombe AGC family kinase Gad8p forms a conserved signaling module with TOR and PDK1-like kinases. EMBO J. 22(12): 3073-83. 12805221
Mayer, C., Zhao, J., Yuan, X. and Grummt, I. (2004). mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 18: 423-434. 15004009
McNeill, H., Craig, G. M. and Bateman, J. M. (2008). Regulation of neurogenesis and epidermal growth factor receptor signaling by the Insulin receptor/Target of rapamycin pathway in Drosophila. Genetics 179: 843-853. PubMed Citation: 18505882
Nicklin, P., et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136(3): 521-34. PubMed Citation: 19203585
Ma, X. M. and Blenis, J. (2009). Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10: 307-318. PubMed ID: 19339977
Migeon, J. C., Garfinkel, M. S. and Edgar, B. A. (1999). Cloning and characterization of peter pan, a novel Drosophila gene required for larval growth. Mol. Biol. Cell 10: 1733-1744. Medline abstract: 10359593
Mohapatra, B., Zutshi, N., An, W., Goetz, B., Arya, P., Bielecki, T. A., Mustaq, I., Storck, M. D., Meza, J. L., Band, V. and Band, H. (2017). An essential role of CBL and CBL-B ubiquitin ligases in mammary stem cell maintenance. Development [Epub ahead of print]. PubMed ID: 28100467
Monserrate, J. P., Chen, M. Y. and Brachmann, C. B. (2012). Drosophila larvae lacking the bcl-2 gene, buffy, are sensitive to nutrient stress, maintain increased basal target of rapamycin (Tor) signaling and exhibit characteristics of altered basal energy metabolism. BMC Biol. 10: 63. PubMed Citation: 22824239
Mori, H., et al. (2000). 14-3-3tau associates with a translational control factor FKBP12-rapamycin-associated protein in T-cells after stimulation by pervanadate. FEBS Lett. 467: 61-64. PubMed Citation: 10664457
Nojima, H., et al. (2003). The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 278(18): 15461-4. 12604610
Oldham, S., Boehni, R., Stocker, H., Brogiolo, W., and Hafen, E. (2000a). Genetic control of size in Drosophila. Phil. Trans. R. Soc. Lond. B 355: 945-952. 11128988
Oldham, S., et al. (2000b). Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 14: 2689-2694. 0523807
Pallares-Cartes, C., Cakan-Akdogan, G. and Teleman, A. A. (2012). Tissue-specific coupling between insulin/IGF and TORC1 signaling via PRAS40 in Drosophila. Dev. Cell 22(1): 172-82. PubMed Citation: 22264732
Panchaud, N., Peli-Gulli, M. P. and De Virgilio, C. (2013). Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal 6: ra42. PubMed ID: 23716719
Parekh, D., et al. (1999). Mammalian TOR controls one of two kinase pathways acting upon nPKCdelta and nPKCepsilon. J. Biol. Chem. 274: 34758-34764. PubMed Citation: 10574945
Parisi, F., et al. (2011). Drosophila insulin and target of rapamycin (TOR) pathways regulate GSK3 beta activity to control Myc stability and determine Myc expression in vivo. BMC Biol. 9: 65. PubMed Citation: 21951762
Park, K. K., Liu, K., Hu, Y., Smith, P. D., Wang, C., Cai, B., Xu, B., Connolly, L., Kramvis, I., Sahin, M. and He, Z. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322: 963-966. PubMed ID: 18988856
Patel, P. H., et al. (2003). Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J. Cell Sci. 116(Pt 17): 3601-10. 12893813
Pelkmans, L., Fava, E., Grabner, H., Hannus, M., Habermann, B., Krausz, E. and Zerial, M. (2005). Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 436: 78-86. Medline abstract: 15889048
Peng, M., Yin, N. and Li, M. O. (2014). Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 159: 122-133. PubMed ID: 25259925
Penney, J., Tsurudome, K., Liao, E. H., Elazzouzi, F., Livingstone, M., Gonzalez, M., Sonenberg, N. and Haghighi, A. P. (2012). TOR is required for the retrograde regulation of synaptic homeostasis at the Drosophila neuromuscular junction. Neuron 74: 166-178. PubMed ID: 22500638
Peña-Llopis, S., Vega-Rubin-de-Celis, S., Schwartz, J. C., Wolff, N. C., Tran, T. A. T., Zou, L., Xie, X.-J., Corey, D. R. and Brugarolas, J. (2011). Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 30: 3242-3258. PubMed ID: 21804531
Peterson, T. R., et al. (2009). DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137(5): 873-86. PubMed Citation: 19446321
Polymenis, M. and Schmidt, E. V. (1997). Coupling of cell division to cell growth by translational control of the G1 cyclin CLN3 in yeast. Genes Dev. 11: 2522-2531. PubMed Citation: 9334317
Powers, T. and Walter, P. (1999). Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 10: 987-1000. PubMed Citation: 10198052
Powers, R. W., et al. (2005). Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20(2): 174-84. Medline abstract: 16418483
Pritchett, T. L. and McCall, K. (2012). Role of the insulin/Tor signaling network in starvation-induced programmed cell death in Drosophila oogenesis. Cell Death Differ. 19(6): 1069-79. PubMed Citation: 22240900
Reddy, P., Zheng, W. and Liu, K. (2010). Mechanisms maintaining the dormancy and survival of mammalian primordial follicles. Trends Endocrinol. Metab. 21: 96-103. PubMed Citation: 19913438
Resnik-Docampo, M. and de Celis, J. F. (2011). MAP4K3 is a component of the TORC1 signalling complex that modulates cell growth and viability in Drosophila melanogaster. PLoS One. 6(1): e14528. PubMed Citation: 21267071
Romero-Pozuelo, J., Demetriades, C., Schroeder, P. and Teleman, A. A. (2017). CycD/Cdk4 and discontinuities in Dpp signaling activate TORC1 in the Drosophila wing disc. Dev Cell 42(4): 376-387 e375. PubMed ID: 28829945
Rong, Y., et al. (2011). Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. 108(19): 7826-31. PubMed Citation: 21518918
Rust, K., Byrnes, L. E., Yu, K. S., Park, J. S., Sneddon, J. B., Tward, A. D. and Nystul, T. G. (2020). A single-cell atlas and lineage analysis of the adult Drosophila ovary. Nat Commun 11(1): 5628. PubMed ID: 33159074
Ruf, V., Holzem, C., Peyman, T., Walz, G., Blackwell, T. K. and Neumann-Haefelin, E. (2013). TORC2 signaling antagonizes SKN-1 to induce C. elegans mesendodermal embryonic development. Dev Biol 384: 214-227. PubMed ID: 23973804
Sabatini, D.., et al. (1994). RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78: 35-43. 7518356
Sancak, Y., et al. (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase Mol. Cell 25: 903-915. PubMed Citation: 17386266
Sancak, Y., Peterson, T. R., Shaul, Y. D., Lindquist, R. A., Thoreen, C. C., Bar-Peled, L. and Sabatini, D. M. (2008). The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496-1501. PubMed ID: 18497260
Sarbassov, D. D., et al. (2004), Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14: 1296-1302. 15268862
Sarbassov, D., Guertin, D. A., Ali, S. M. and Sabatini, D. M. (2005a). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098-1101. Medline abstract: 15718470
Sarbassov, D. D. and Sabatini, D. M. (2005b). Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 280(47): 39505-9. Medline abstract: 16183647
Sarbassov, D. D., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22(2): 159-68. Medline abstract: 16603397
Saucedo, L. J., et al. (2003). Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 5(6):566-71. 12766776
Schalm, S. S. and Blenis, J. (2002). Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12(8): 632-9. 11967149
Schalm, S. S., Fingar, D. C., Sabatini, D. M. and Blenis, J. (2003). TOS motif-mediated Raptor Binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 13(10): 797-806. 12747827
Schieber, M. and Chandel, N. S. (2014). TOR signaling couples oxygen sensing to lifespan in C. elegans. Cell Rep 9: 9-15. PubMed ID: 25284791
Scott, P. H., et al. (1999). Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc. Natl. Acad. Sci. 95: 7772-7777. 9636226
Scott, R. C., Schuldiner, O. and Neufeld, T. P. (2004). Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 7: 167-178. 15296714
Scott, R. C., Juhász, G. and Neufeld, T. P. (2007). Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 17(1): 1-11. PubMed Citation: 17208179
Senger, S., Csokmay, J., Akbar, T., Jones, T. I., Sengupta, P. and Lilly, M. A. (2011).The nucleoporin Seh1 forms a complex with Mio and serves an essential tissue-specific function in Drosophila oogenesis. Development 138(10): 2133-42. PubMed ID: 21521741
Settembre, C., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332: 1429-1433. PubMed ID: 21617040
Shah, O.J., Wang, Z. and Hunter. T. (2004). Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 14: 1650-1656. Medline abstract: 15380067
Shen, K., Sidik, H. and Talbot, W. S. (2016). The Rag-Ragulator complex regulates lysosome function and phagocytic flux in microglia. Cell Rep 14: 547-559. PubMed ID: 26774477
Sheaffer, K. L., Updike, D. L. and Mango, S. E. (2008). The Target of Rapamycin pathway antagonizes pha-4/FoxA to control development and aging. Curr. Biol. 18(18): 1355-64. PubMed Citation: 18804378
Shibutani, S. T. et al. (2008). Intrinsic negative cell cycle regulation provided by PIP box- and Cul4Cdt2-mediated destruction of E2f1 during S phase. Dev. Cell 15: 890-900. PubMed Citation: 19081076
Sinagoga, K. L., Stone, W. J., Schiesser, J. V., Schweitzer, J. I., Sampson, L., Zheng, Y. and Wells, J. M. (2017). Distinct roles for the mTOR pathway in postnatal morphogenesis, maturation and function of pancreatic islets. Development [Epub ahead of print]. PubMed ID: 28576773
Slack, C., Giannakou, M. E., Foley, A., Goss, M. and Partridge, L. (2011). dFOXO-independent effects of reduced insulin-like signaling in Drosophila. Aging Cell 10: 735-748. PubMed ID: 21443682
Sofer, A., Lei, K., Johannessen, C. M. and Ellisen, L. W. (2005). Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 25(14): 5834-45. 15988001
Sonenberg, N. and Hinnebusch, A. G. (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136: 731-745. PubMed ID: 19239892
Soukas, A. A., Kane, E. A., Carr, C. E., Melo, J. A. and Ruvkun, G. (2009). Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev. 23(4): 496-511. PubMed Citation: 19240135
Sousa-Nunes, R., Yee, L. L. and Gould, A. P. (2011). Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature 471(7339): 508-12. PubMed Citation: 21346761
Steiger, D., Furrer, M., Schwinkendorf, D. and Gallant, P. (2008). Max-independent functions of Myc in Drosophila melanogaster. Nat. Genet. 40(9): 1084-1091. PubMed Citation: 19165923
Stocker, H., et al. (2003). Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat. Cell Biol. 5(6):559-65. 12766775
Sun, P., et al. (2010). TSC1/2 tumour suppressor complex maintains Drosophila germline stem cells by preventing differentiation. Development 137: 2461-2469. PubMed Citation: 20573703
Sutton, M. A. and Schuman, E. M. (2006). Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127: 49-58. PubMed ID: 17018276
Swiech, L., Blazejczyk, M., Urbanska, M., Pietruszka, P., Dortland, B. R., Malik, A. R., Wulf, P. S., Hoogenraad, C. C. and Jaworski, J. (2011). CLIP-170 and IQGAP1 cooperatively regulate dendrite morphology. J Neurosci 31(12): 4555-4568. PubMed ID: 21430156
Texada, M. J., Lassen, M., Pedersen, L. H., Koyama, T., Malita, A. and Rewitz, K. (2022). Insulin signaling couples growth and early maturation to cholesterol intake in Drosophila. Curr Biol 32(7): 1548-1562. PubMed ID: 35245460
Tiebe, M., Lutz, M., De La Garza, A., Buechling, T., Boutros, M. and Teleman, A. A. (2015). Reptor and Reptor-BP regulate organismal metabolism and transcription downstream of TORC1. Dev Cell 33: 272-284. PubMed ID: 25920570
Tang, S. J., et al. (2002). A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. 99(1): 467-72. 11756682
Tee, A. R., et al. (2003). Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 13(15): 1259-68. 12906785
Teleman, A. A., Chen, Y. W. and Cohen, S. M. (2005). Drosophila Melted modulates FOXO and TOR activity. Dev Cell 9(2): 271-81. 16054033
Teleman, A. A., Hietakangas, V., Sayadian, A. C. and Cohen, S. M. (2008). Nutritional control of protein biosynthetic capacity by insulin via Myc in Drosophila. Cell Metab 7: 21-32. PubMed ID: 18177722
Teotia, P., Van Hook, M. J., Fischer, D. and Ahmad, I. (2019). Human retinal ganglion cell axon regeneration by recapitulating developmental mechanisms: effects of recruitment of the mTOR pathway. Development 146(13). PubMed ID: 31273087
Thomas, G. and Hall, M. N. (1997). TOR signaling and control of cell growth. Curr. Opin. Cell Biol. 9: 782-787. 9425342
Thomas, G. (2002). The S6 kinase signaling pathway in the control of development and growth. Biol. Res. 35: 305-313. 12415748
Thomson, T. C., Fitzpatrick, K. E. and Johnson, J. (2010). Intrinsic and extrinsic mechanisms of oocyte loss. Mol. Hum. Reprod. 16: 916-927. PubMed Citation: 20651035
Topalidou, I., Papamichos-Chronakis, M. and Thireos, G. (2003). Post-TATA binding protein recruitment clearance of Gcn5-dependent histone acetylation within promoter nucleosomes. Mol. Cell. Biol. 23(21): 7809-17. 14560024
Tsang, C. K., Bertram, P. G., Ai, W., Drenan, R. and Zheng, X. F. (2003). Chromatin-mediated regulation of nucleolar structure and RNA Pol I localization by TOR. EMBO J. 22(22): 6045-56. 14609951
Vander Haar, R., et al. (2007). Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9: 316-323. PubMed Citation: 17277771
Venkatachalam, K., Long, A. A., Elsaesser, R., Nikolaeva, D., Broadie, K. and Montell, C. (2008). Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell 135: 838-851. PubMed ID: 19041749
Vachias, C., Fritsch, C., Pouchin, P., Bardot, O. and Mirouse, V. (2014). Tight coordination of growth and differentiation between germline and soma provides robustness for Drosophila egg development. Cell Rep 9: 531-541. ID: PubMed
Wang, L, Fraley, C. D., Faridi, J., Kornberg, A. and Roth. R. A. (2003). Inorganic polyphosphate stimulates mammalian TOR, a kinase involved in the proliferation of mammary cancer cells. Proc. Natl. Acad. Sci. 100(20) :11249-54. 12970465
Wang, Y. H. and Huang, M. L. (2009). Reduction of Lobe leads to TORC1 hypoactivation that induces ectopic Jak/STAT signaling to impair Drosophila eye development. Mech. Dev. 126(10): 781-790. PubMed Citation: 19733656
Wang, T., Blumhagen, R., Lao, U., Kuo, Y. and Edgar, B. A. (2012). LST8 regulates cell growth via target-of-rapamycin complex 2 (TORC2). Mol Cell Biol 32: 2203-2213. PubMed ID: 22493059
Wassarman, D. R., Bankapalli, K., Pallanck, L. J. and Shokat, K. M. (2022). Tissue-restricted inhibition of mTOR using chemical genetics. Proc Natl Acad Sci U S A 119(38): e2204083119. PubMed ID: 36095197
Wei, Y. and Lilly, M. A. (2014a). The TORC1 inhibitors Nprl2 and Nprl3 mediate an adaptive response to amino-acid starvation in Drosophila. Cell Death Differ 21: 1460-1468. PubMed ID: 24786828
Wei, Y., Reveal, B., Reich, J., Laursen, W. J., Senger, S., Akbar, T., Iida-Jones, T., Cai, W., Jarnik, M. and Lilly, M. A. (2014b). TORC1 regulators Iml1/GATOR1 and GATOR2 control meiotic entry and oocyte development in Drosophila. Proc Natl Acad Sci U S A 111(52):E5670-7. PubMed ID: 25512509
Wei, Y., Bettedi, L., Ting, C. Y., Kim, K., Zhang, Y., Cai, J. and Lilly, M. A. (2019). The GATOR complex regulates an essential response to meiotic double-stranded breaks in Drosophila. Elife 8. PubMed ID: 31650955
Wieczorek, H., Beyenbach, K. W., Huss, M. and Vitavska, O. (2009). Vacuolar-type proton pumps in insect epithelia. J. Exp. Biol. 212: 1611-1619. PubMed ID: 19448071
Wittmann, C. W., et al. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:. 711-714. 11408621
Wong, C. O., Li, R., Montell, C. and Venkatachalam, K. (2012). Drosophila TRPML is required for TORC1 activation. Curr Biol 22: 1616-1621. PubMed ID: 22863314
Yang, G., Humphrey, S. J., Murashige, D. S., Francis, D., Wang, Q. P., Cooke, K. C., Neely, G. and James, D. E. (2018). RagC phosphorylation autoregulates mTOR complex 1. EMBO J. PubMed ID: 30552228
Yokogami, K., et al. (2000). Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 10: 47-50. PubMed Citation: 10660304
Zaffran, S., et al. (1998). A Drosophila RNA helicase gene, pitchoune, is required for cell growth and proliferation and is a potential target of d-Myc. Development 125: 3571-3584. Medline abstract: 9716523
Zaragoza, D., et al. (1998). Rapamycin induces the G0 program of transcriptional repression in yeast by interfering with the TOR signaling pathway. Mol. Cell. Biol. 18: 4463-4470. PubMed Citation: 9671456
Zeng, Z., et al. (2006). Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 109(8): 3509-12. Medline abstract: 17179228
Zhang, H., et al. (2000). Regulation of cellular growth by the drosophila target of rapamycin dTOR. Genes Dev. 14(21): 2712-24. PubMed Citation: 11069888
Zhang, T., Zhou, Q., Ogmundsdottir, M. H., Moller, K., Siddaway, R., Larue, L., Hsing, M., Kong, S. W., Goding, C. R., Palsson, A., Steingrimsson, E. and Pignoni, F. (2015). Mitf is a master regulator of the v-ATPase, forming a control module for cellular homeostasis with v-ATPase and TORC1. J Cell Sci 128: 2938-2950. PubMed ID: 26092939
Zhang, Y., et al. (2003). Rheb is a direct target of the tuberous sclerosis tumor suppressor proteins. Nat. Cell Biol. 5(6): 578-81. 12771962
Zheng, X. and Sehgal, A. (2010). AKT and TOR signaling set the pace of the circadian pacemaker. Curr. Biol. 20(13): 1203-8. PubMed Citation: 20619819
Zielke, N., et al. (2011). Control of Drosophila endocycles by E2F and CRL4CDT2. Nature 480(7375): 123-7. PubMed Citation: 22037307
Zinke, I., et al. (2002). Nutrient control of gene expression in Drosophila: microarray analysis of starvation and sugar-dependent response. EMBO J. 21: 6162-6173. Medline abstract: 12426388
Zoncu, R., Bar-Peled, L., Efeyan, A., Wang, S., Sancak, Y. and Sabatini, D. M. (2011). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334: 678-683. PubMed ID: 22053050
date revised: 25 January 2026
Home page: The Interactive Fly © 2026 Thomas Brody, Ph.D.
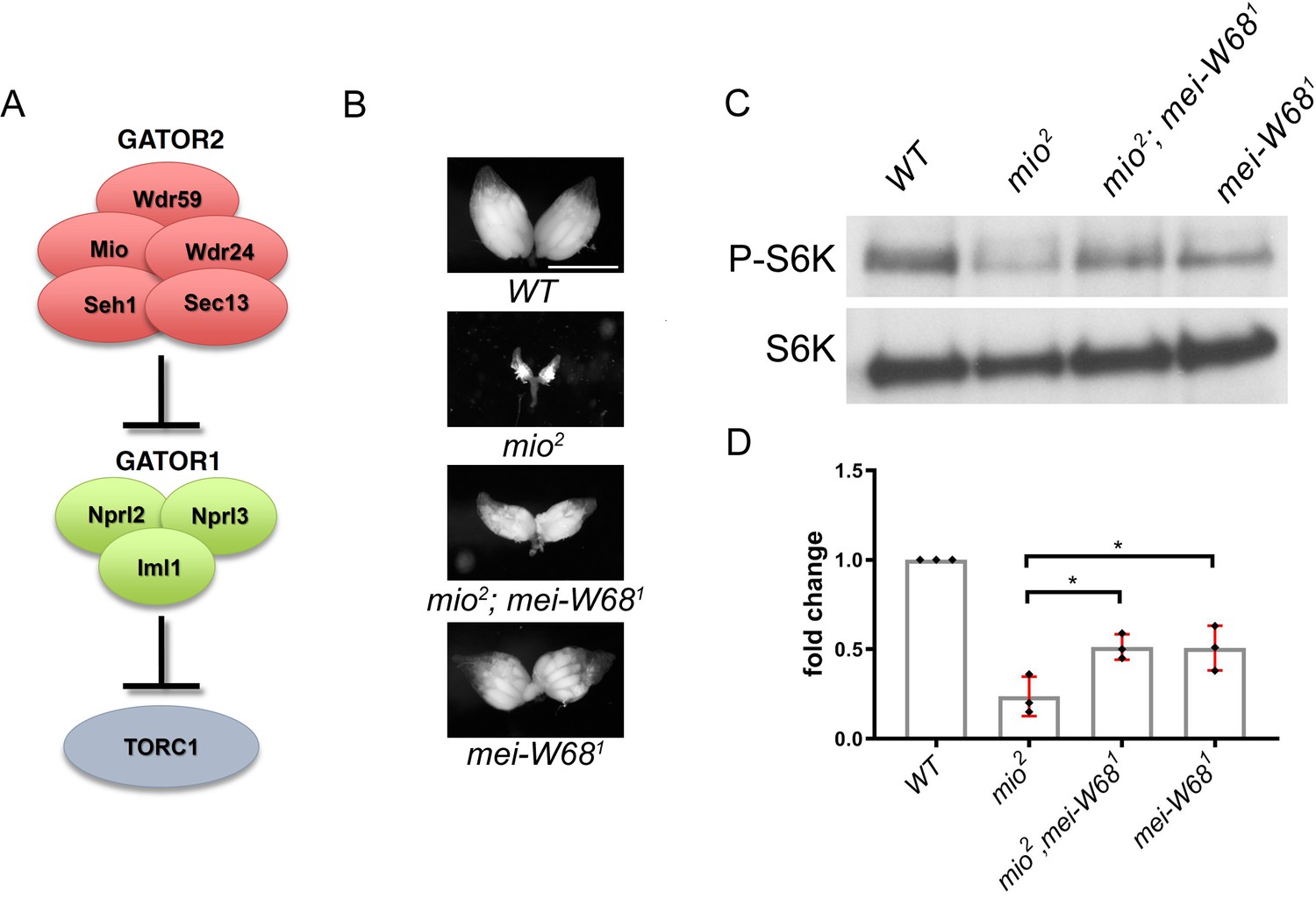{kind=link}
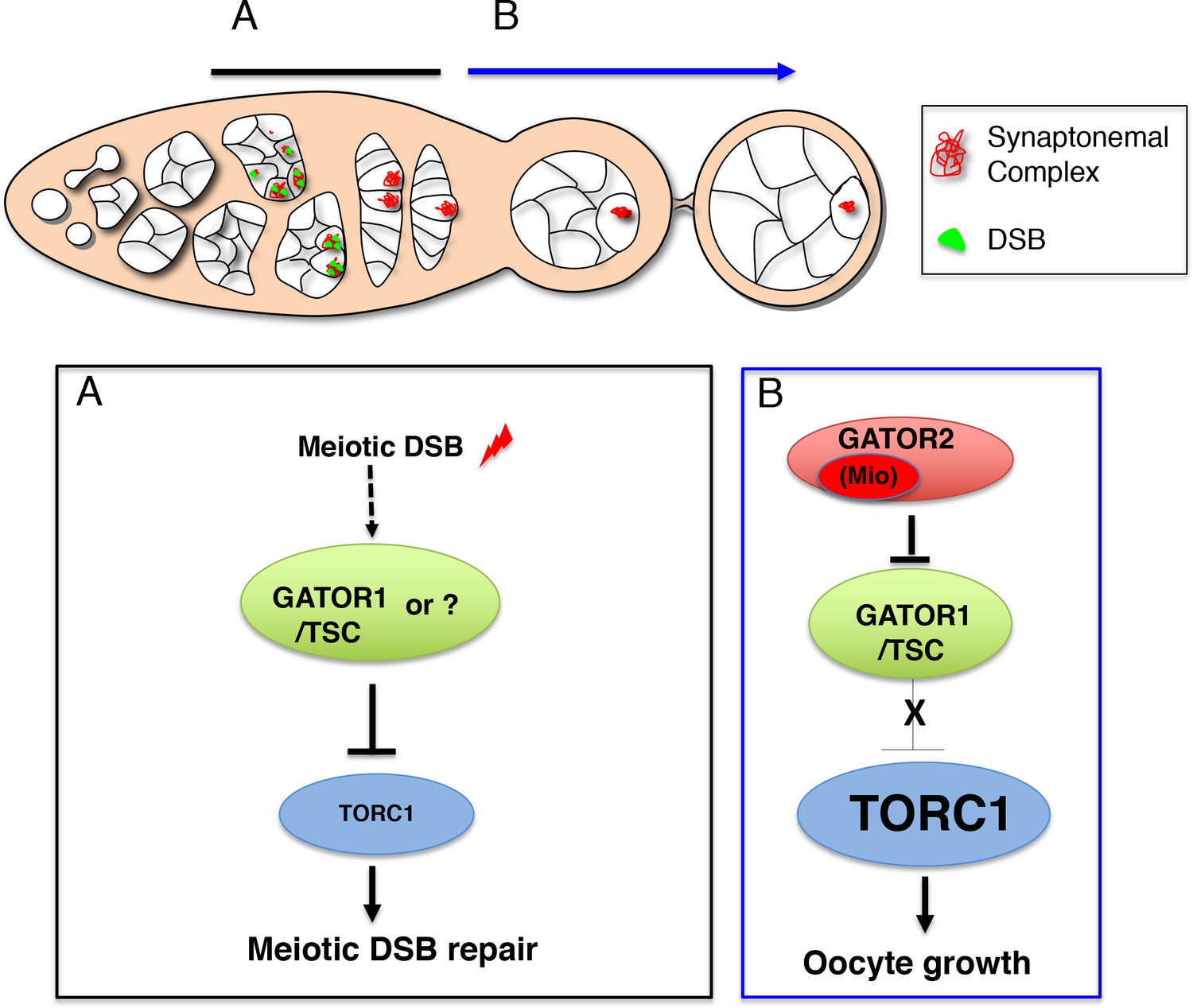{kind=link}